震撼发布!青蒿素再亮剑!中国科学技术大学团队解密"东方神药"新功效:青蒿素衍生物SM934靶向代谢酶逆转肥胖相关炎症! 2026年5月19日
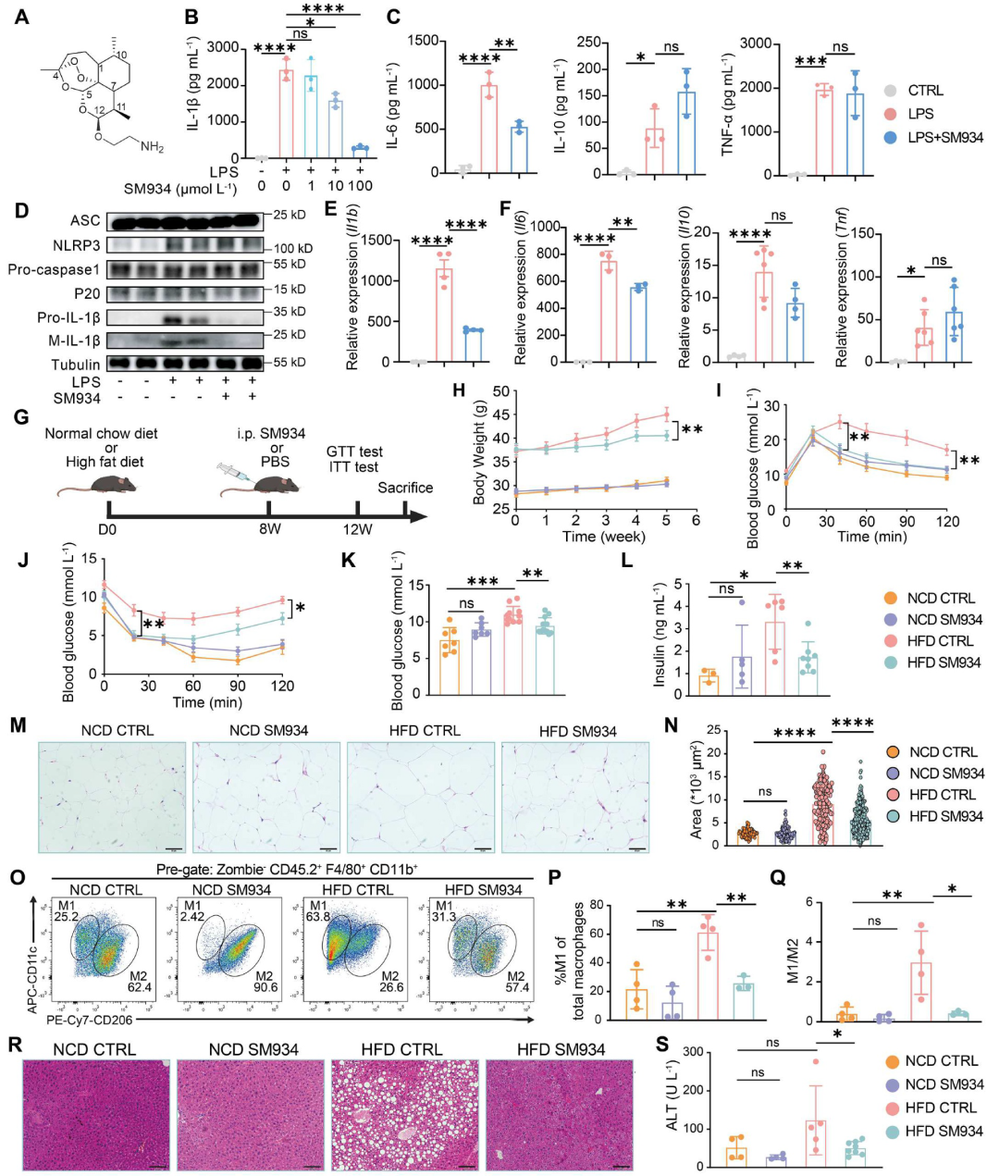
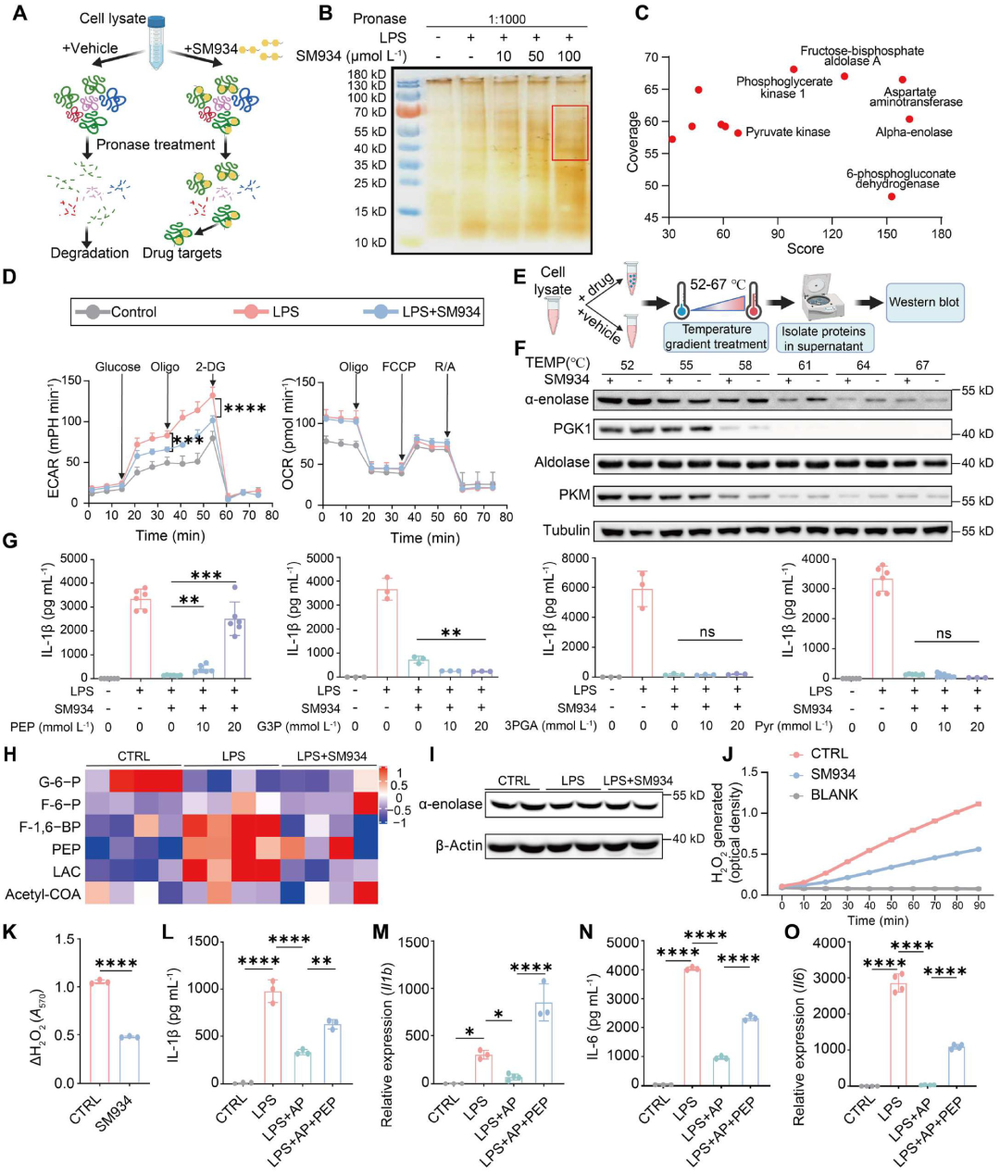
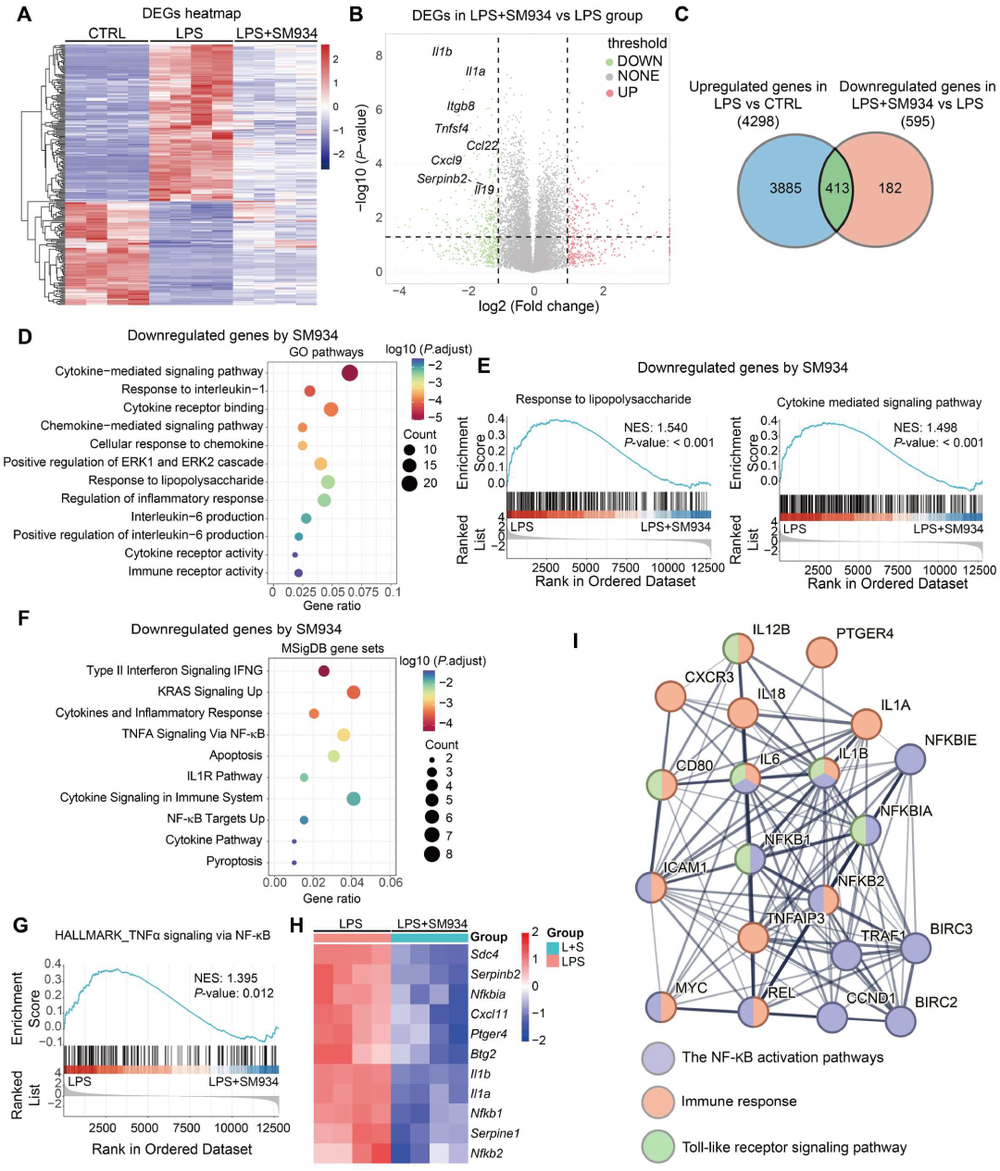
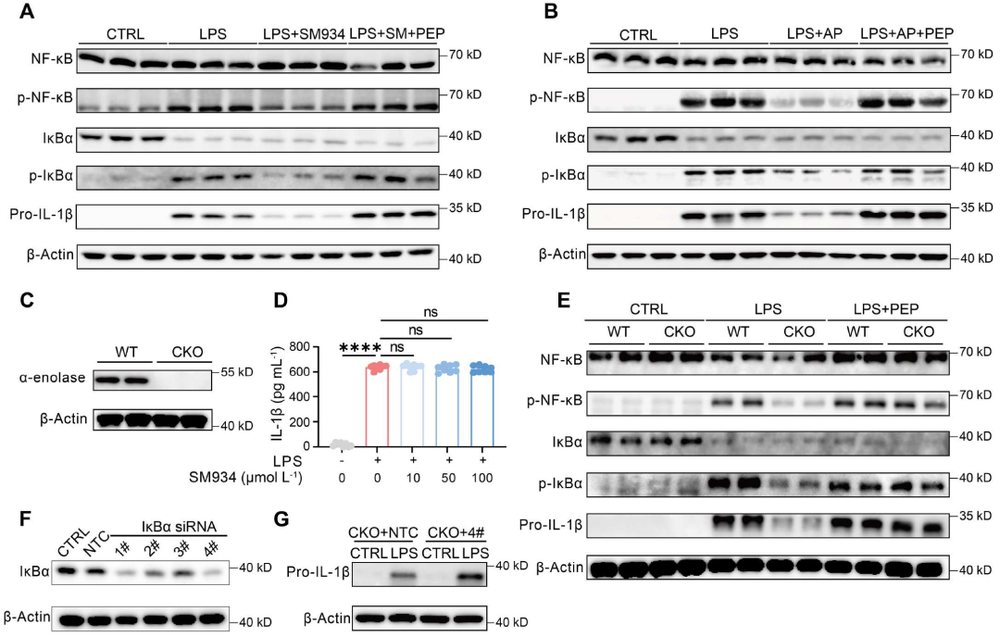
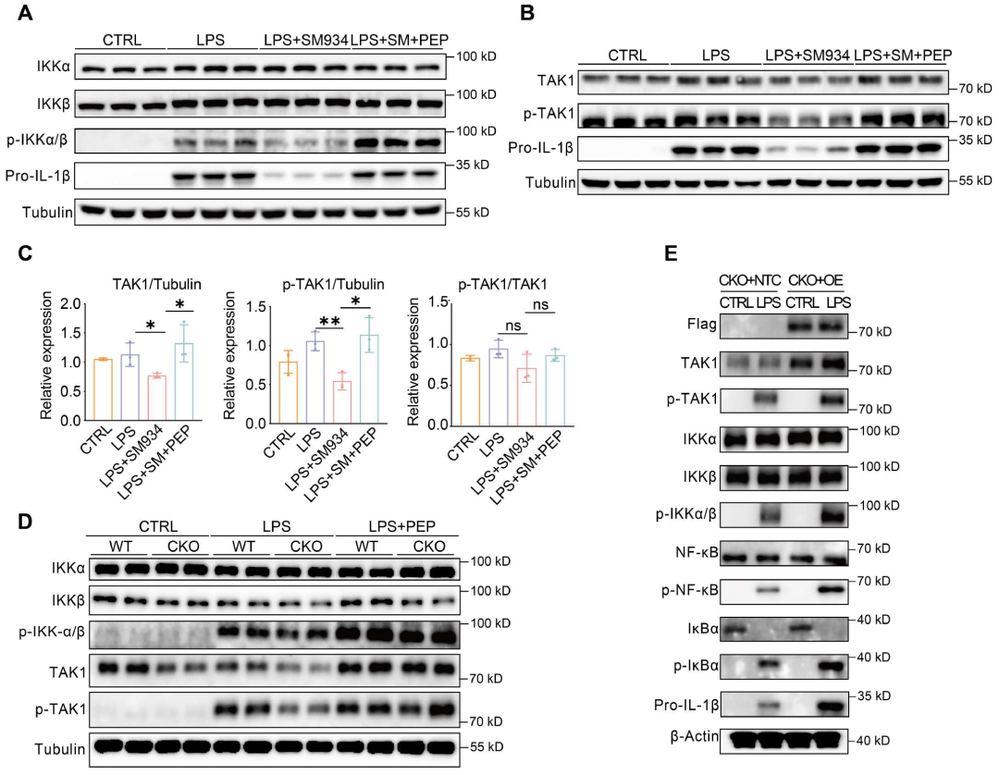
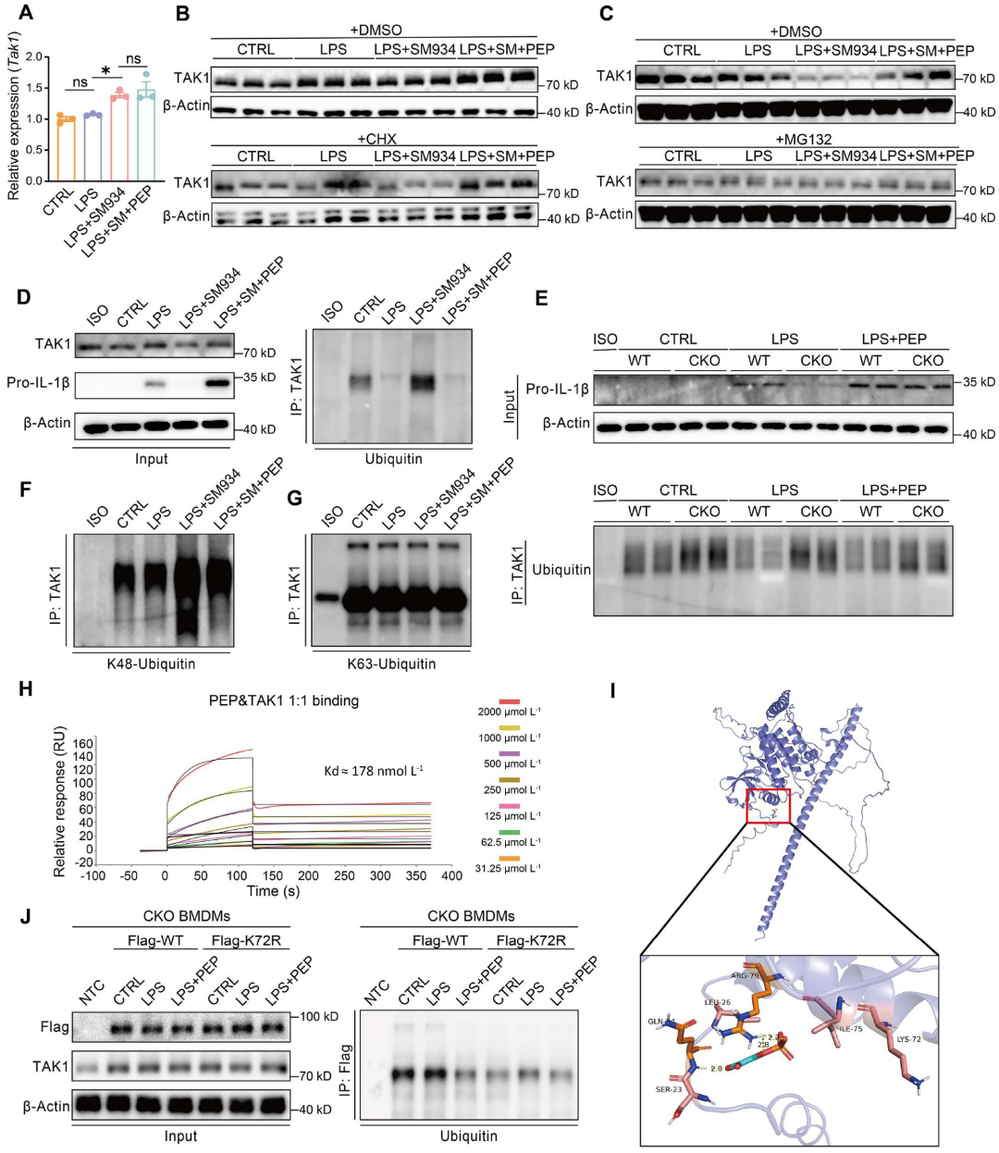
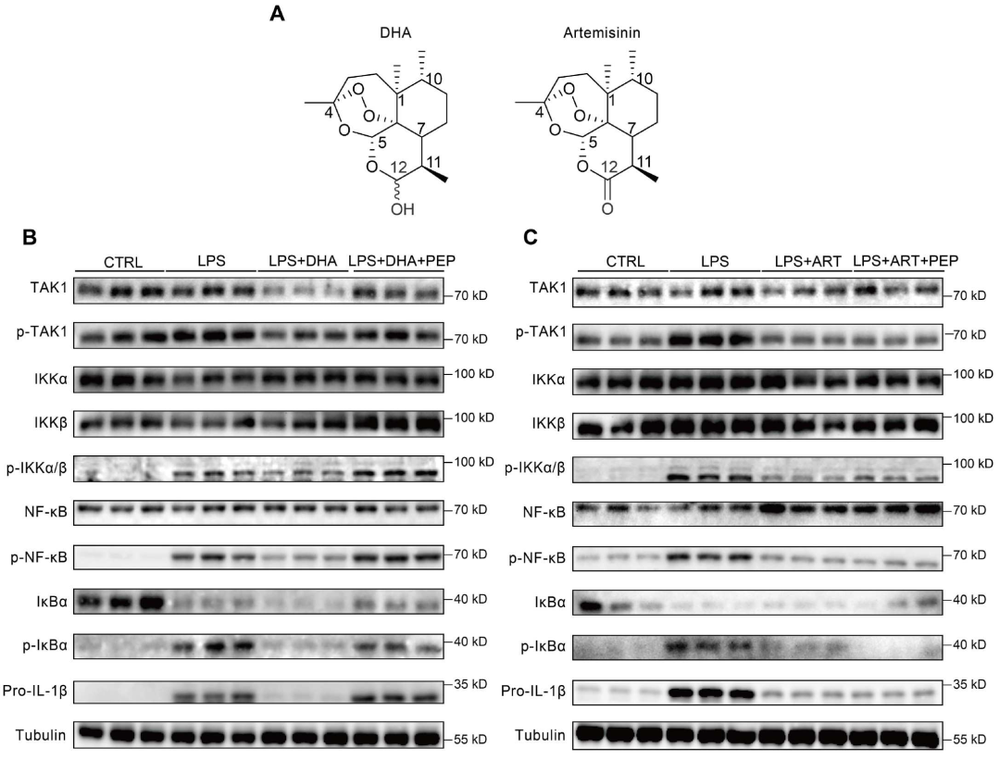
炎症 是机体抵御威胁的一种防御形式,然而慢性炎症已与多种疾病密切相关,包括心血管疾病、代谢性疾病、神经退行性疾病、自身免疫性疾病以及癌症。转化生长因子-β激活的激酶1(TAK1)是MAPK激酶激酶(MAPKKK)家族成员,已被报道为炎症反应中的关键信号枢纽。TAK1可被TLR配体、TNF-α和IL-1β激活,进而激活下游的NF-κB和MAPK信号通路。近年来,代谢与免疫功能的关联受到广泛关注,代谢产物能够通过表观遗传调控以及与信号蛋白的共价或非共价结合来控制炎症细胞反应,但细胞代谢与TAK1通路之间的串扰仍不清楚,代谢物如何调控TAK1激活并作为药物靶点仍有待探索。 目前主要的抗炎药物包括糖皮质激素和非甾体抗炎药,但由于其不良反应,具有良好抗炎活性的天然产物备受关注。 青蒿素 是从中药青蒿中发现的传统药物,已用于治疗疟疾数百年,近年来青蒿素及其衍生物的抗炎作用逐渐被揭示。其中, 水溶性青蒿素衍生物β-氨基青蒿醚马来酸盐(SM934) 已被报道为一种良好的免疫调节剂,能够干扰先天免疫应答和适应性免疫应答。 近期,一项研究深入探讨了SM934调控炎症反应的分子机制,为青蒿素类药物的临床应用提供了新的理论基础。 青蒿素衍生物SM934抑制巨噬细胞IL-1β反应并减轻代谢性炎症性疾病 研究人员首先系统评估了SM934对巨噬细胞炎症反应的影响,发现该化合物能够以剂量依赖的方式显著抑制脂多糖联合三磷酸腺苷刺激后骨髓来源巨噬细胞中白介素-1β的产生,在100微摩尔每升浓度下抑制效果最为显著,同时对白介素-6的产生也有一定程度的降低,但不影响白介素-10和肿瘤坏死因子-α的分泌水平。进一步的蛋白质免疫印迹分析揭示, SM934并不干扰炎症小体的正常激活过程,即不影响凋亡相关斑点样蛋白、NLRP3、pro-caspase-1和caspase-1亚基p20的表达水平,而是通过降低pro-IL-1β的蛋白合成来发挥作用 ,这与该化合物显著下调Il1b基因转录水平的结果完全一致 (图1D) 。上述现象在小鼠腹腔巨噬细胞和人单核细胞来源巨噬细胞中均得到验证,表明SM934对IL-1β反应的抑制作用具有物种保守性和细胞类型普遍性。 鉴于IL-1β在肥胖相关代谢性炎症中的核心驱动作用,研究团队进一步利用高脂饮食诱导的肥胖小鼠模型评估了SM934的体内药效。实验设计上,小鼠先接受正常饲料或高脂饲料喂养8周,随后再给予SM934或磷酸盐缓冲液处理4周,结果显示SM934显著降低了高脂饮食小鼠的体重增长,改善了葡萄糖耐量和胰岛素敏感性,降低了空腹血糖和空腹胰岛素水平,而对正常饮食小鼠的代谢指标无明显影响。组织病理学分析表明, SM934处理显著缩小了白色脂肪组织中脂肪细胞的体积,抑制了脂肪细胞肥大 (图1R) 。 流式细胞术分析进一步揭示,SM934显著降低了脂肪组织中促炎型M1巨噬细胞的比例,同时增加了抗炎型M2巨噬细胞的比例,使得M1/M2比值明显下降,提示脂肪组织代谢性炎症得到有效缓解 (图1Q) 。此外,SM934还显著改善了高脂饮食诱导的肝脏脂肪变性和损伤,血清丙氨酸氨基转移酶水平明显降低 。 这些体内外实验结果共同证实,SM934能够选择性抑制巨噬细胞IL-1β反应并有效减轻代谢性炎症性疾病的病理进展。 SM934通过靶向α-烯醇化酶并抑制PEP产生来抑制IL-1β反应 为了鉴定SM934在巨噬细胞中的潜在靶点,研究人员采用了药物亲和反应靶点稳定性(DARTS)方法。该方法基于配体结合增加蛋白质对蛋白水解的抗性原理(图2A)。将BMDM裂解液与SM934孵育后,用pronase处理,从凝胶中回收抗蛋白水解的蛋白质并进行质谱鉴定(图2B)。结果显示,α-烯醇化酶(α-enolase)是得分最高的蛋白质(图2C),且高分蛋白质功能上与细胞呼吸和糖酵解相关,提示SM934调控细胞代谢。接下来,研究人员使用Seahorse分析仪评估了SM934对BMDMs有氧呼吸和糖酵解能力的影响。如图2D所示, SM934抑制了活化BMDMs的总体糖酵解能力,但不影响有氧呼吸,表明SM934靶向糖酵解相关分子。 为进一步验证SM934与糖酵解酶的相互作用,研究人员采用了细胞热迁移实验(CETSA)。如图2E、2F所示,随着温度升高,所有蛋白质的上清液水平均因热变性而降低,但只有α-烯醇化酶的水平受到SM934的影响,而磷酸甘油酸激酶1(PGK1)、醛缩酶和丙酮酸激酶肌肉同工酶(PKM)等其他糖酵解相关蛋白的水平未受影响,表明SM934与α-烯醇化酶存在直接结合。研究人员进一步发现,磷酸烯醇式丙酮酸(PEP)能够恢复SM934处理的BMDMs在LPS+ATP刺激后的IL-1β产生,而添加甘油醛-3-磷酸(G3P)、3-磷酸甘油酸(3PGA)和丙酮酸等其他糖酵解中间产物则无明显效果(图2G), 提示SM934通过靶向α-烯醇化酶并抑制PEP产生来抑制IL-1β反应 。 非靶向代谢组学分析证实,SM934降低了LPS刺激BMDMs中的PEP水平(图2H)。Western blot分析显示,SM934不影响总α-烯醇化酶蛋白的表达(图2I),但酶活性测定表明SM934能够有效抑制α-烯醇化酶活性(图2J、2K)。此外,使用已知的烯醇化酶抑制剂AP-3-A4(AP)处理LPS+ATP刺激的BMDMs,发现其抑制了IL-1β产生并降低了Il1b mRNA水平,而这些抑制作用可通过补充PEP得到挽救(图2L、2M)。AP-3-A4对IL-6在蛋白和mRNA水平的表达也有相同效果(图2N、2O)。 这些数据共同证实,SM934通过干扰α-烯醇化酶活性来抑制巨噬细胞的IL-1β反应。 SM934抑制巨噬细胞中的炎症通路和NF-κB激活 为进一步揭示SM934通过减少PEP抑制IL-1β产生的潜在机制,研究人员对未刺激BMDMs、LPS刺激BMDMs以及SM934处理的LPS刺激BMDMs进行了转录组分析。基因表达谱分析显示,LPS改变了BMDMs的转录景观,而SM934部分恢复了这一改变(图3A)。特别值得注意的是,SM934下调了与炎症反应相关的基因,包括Il1b、Il1a和Cxcl9(图3B)。在LPS上调的基因中,有413个基因被SM934下调(图3C),GO分析显示这些基因与IL-1反应、趋化因子受体结合和Toll样受体信号通路等炎症相关信号通路有关(图3D)。基因集富集分析(GSEA)进一步富集到对脂多糖的反应和细胞因子介导的信号通路(图3E)。利用MSigDB数据库进行深度富集分析,研究人员在这413个差异表达基因中鉴定出NF-κB通路(图3F、3G)。 NF-κB是炎症反应中关键的转录因子,SM934处理降低了LPS刺激BMDMs中NF-κB通路相关基因的表达 (图3H)。此外, 基因串扰分析显示NF-κB通路在调控TLR信号和免疫反应相关基因中发挥核心作用 (图3I)。 与转录组数据一致,Western blot分析显示,与LPS刺激的BMDMs相比,SM934处理的LPS刺激BMDMs中pro-IL-1β水平降低,IκBα和NF-κB的磷酸化水平也降低(图4A),表明SM934损害了NF-κB通路的激活。PEP补充恢复了pro-IL-1β的产生以及IκBα和NF-κB的磷酸化(图4A),表明PEP是NF-κB通路激活所必需的。同样,α-烯醇化酶抑制剂AP-3-A4抑制了LPS刺激BMDMs中pro-IL-1β的产生以及IκBα和NF-κB的磷酸化,而这些抑制作用可通过PEP补充恢复(图4B)。 为进一步证明PEP在激活NF-κB通路中的作用,研究人员构建了Lyz2cre Eno1f/f小鼠,特异性敲除髓系细胞中的α-烯醇化酶(图4C)。结果显示,SM934未能抑制α-烯醇化酶缺陷BMDMs中IL-1β的产生(图4D),证实SM934的作用是通过α-烯醇化酶介导的。此外,LPS刺激的α-烯醇化酶缺陷BMDMs中NF-κB和IκBα的磷酸化以及pro-IL-1β的产生均降低,而这些降低可通过外源PEP补充恢复(图4E)。为进一步确认NF-κB激活受损是PEP缺乏导致IL-1β表达降低的原因,研究人员在Lyz2cre Eno1f/f BMDMs中敲低IκBα以增强NF-κB激活,发现这成功增加了LPS诱导的IL-1β产生(图4F、4G)。 这些数据共同证明,PEP通过激活NF-κB通路促进IL-1β产生。 PEP通过维持TAK1蛋白水平促进TAK1-IKK-NF-κB通路激活 既往研究表明,LPS-TLR4信号触发TAK1-IKK-NF-κB轴的级联激活。为探索PEP促进NF-κB激活的机制,研究人员评估了PEP对IKK激活的影响。如图5A所示,SM934降低了LPS诱导的IKKα/β磷酸化,但不影响其总蛋白水平,而PEP补充恢复了磷酸化IKKα/β水平。IKK磷酸化由TAK1介导,因此研究人员进一步研究了TAK1的激活。如图5B、5C所示,SM934降低了LPS诱导的TAK1磷酸化,而PEP补充恢复了磷酸化TAK1水平。值得注意的是,SM934还降低了LPS刺激BMDMs中总TAK1蛋白水平,PEP补充恢复了TAK1水平(图5B、5C)。重要的是,磷酸化TAK1与总TAK1的比值不受SM934或PEP的影响(图5C),这些数据表明PEP缺乏导致的TAK1磷酸化降低是由于总TAK1蛋白减少,而非磷酸化过程受损。类似地,LPS刺激的Lyz2cre Eno1f/f BMDMs与对照BMDMs相比,IKKα/β和TAK1的磷酸化以及总TAK1蛋白水平均降低,而这些由α-烯醇化酶缺陷引起的变化可通过PEP补充逆转(图5D)。此外,在α-烯醇化酶缺陷BMDMs中过表达TAK1增强了IKKα/β、IκBα和NF-κB的磷酸化(图5E), 证实PEP产生减少通过降低TAK1蛋白水平损害了NF-κB激活。 PEP通过阻碍赖氨酸72位点的泛素化抑制TAK1降解 尽管TAK1蛋白水平降低,但SM934并未降低Tak1的mRNA水平,甚至略有增加(图6A)。此外,PEP对Tak1 mRNA水平也无影响(图6A),这些数据提示PEP在蛋白水平控制TAK1含量。接下来,研究人员探究了PEP是否调控TAK1蛋白降解或蛋白质合成。当用放线菌酮(CHX)抑制蛋白质合成时,SM934在LPS刺激BMDMs中以与未处理CHX的对照细胞相似的方式降低TAK1蛋白水平(图6B)。另一方面,当用蛋白酶体抑制剂MG132处理LPS刺激BMDMs时,SM934对TAK1蛋白水平的影响被消除(图6C), 这些结果表明PEP通过控制蛋白质降解而非蛋白质合成来调控TAK1蛋白水平。 进一步的免疫共沉淀分析显示,SM934处理增加了TAK1的泛素化,而PEP补充降低了TAK1泛素化(图6D)。在LPS刺激的Lyz2cre Eno1f/f BMDMs中,与LPS刺激的对照BMDMs相比,TAK1泛素化也增加,而PEP补充抑制了这一泛素化(图6E)。泛素化主要有两种类型:K48连接和K63连接泛素化,其中K48泛素化调控蛋白质降解。免疫印迹证实, SM934促进了TAK1与K48泛素化而非K63泛素化的结合,而PEP补充减少了TAK1与K48泛素化的结合 (图6F、6G),支持PEP抑制泛素化介导的TAK1降解。 当PEP用于预处理BMDMs然后去除时,未能恢复SM934抑制的IL-1β表达,提示PEP以非共价方式发挥作用。表面等离子体共振(SPR)分析显示,PEP直接与TAK1结合,解离常数(Kd)为178 nmol L⁻¹(图6H)。既往研究报道,赖氨酸-72(K72)位点的K48连接泛素化介导TAK1降解。分子对接分析预测PEP的结合位点靠近赖氨酸-72位点(图6I)。接下来,研究人员探究了PEP是否影响TAK1在赖氨酸-72位点的泛素化。在Lyz2cre Eno1f/f BMDMs中分别过表达野生型(WT)TAK1和TAK1-K72R突变体,K72R突变消除了TAK1泛素化,PEP补充抑制了WT TAK1的泛素化,但对TAK1-K72R突变体无影响(图6J)。 这些数据共同证明,PEP通过干扰TAK1在赖氨酸-72位点的泛素化来抑制TAK1降解。 DHA而非青蒿素通过降低TAK1抑制NF-κB通路激活 既往研究表明青蒿素及其衍生物双氢青蒿素(DHA)能够损害NF-κB通路激活。如图7A所示,DHA和青蒿素的化学结构存在差异,DHA在C12位点具有羟基取代,而青蒿素在C12位点具有羰基。 本研究发现,DHA和青蒿素均能损害NF-κB通路激活并减少BMDMs中pro-IL-1β的产生 (图7B、7C)。然而,只有DHA降低了TAK1蛋白水平,青蒿素不影响TAK1水平。此外,PEP补充恢复了DHA处理BMDMs中的TAK1蛋白水平、NF-κB通路激活和pro-IL-1β产生,但未能恢复青蒿素处理BMDMs中的NF-κB通路激活和pro-IL-1β产生(图7B、7C)。这些结果表明,青蒿素及其衍生物通过不同机制抑制NF-κB通路激活。SM934在C1位点缺乏甲基,降低了其亲脂性同时增加了水溶性。与青蒿素相比,DHA和SM934在C12位点分别具有羟基或2-氨基乙氧基取代而非原始的碳基团,这些结构修饰引入了额外的氢键供体,可能通过增加氢键结合能力增强与氨基酸的分子相互作用,从而促进非共价结合。 这些结构差异可能导致青蒿素及其衍生物具有不同的作用机制。 研究揭示了青蒿素衍生物SM934抑制炎症反应的全新分子机制。SM934直接靶向糖酵解酶α-烯醇化酶,抑制其酶活性,从而减少代谢产物PEP的产生。PEP直接与TAK1结合,以非共价方式干扰TAK1在赖氨酸-72位点的K48连接泛素化,抑制蛋白酶体介导的TAK1降解,维持TAK1蛋白稳定性。稳定的TAK1蛋白保证了TAK1-IKK-NF-κB信号轴的级联激活,促进NF-κB通路活化,进而驱动IL-1β和IL-6等炎症因子的产生。 这一发现首次阐明了PEP介导的TAK1代谢调控机制,将细胞糖酵解代谢与先天免疫炎症反应紧密联系起来,为青蒿素类药物在炎症性疾病和代谢性疾病中的应用提供了坚实的理论基础。
参考文献: Pan J, Zhang Y, Du Z, Huang Y, Wang Y, Su M, Cai Y, Xu W, Chen L, Huang C, Cui D, Chen D, Fu S, Wu Q, Tian C, Tang M, Ji T, Hou J, Zuo J, Li S, Zhang H, Bai L. Artemisinin derivative SM934 targets α-enolase to inhibit PEP-mediated TAK1 stabilization and inflammation. Sci China Life Sci. 2026 Apr 17. doi: 10.1007/s11427-025-3167-7. Epub ahead of print. PMID: 42024180. 老中医(微信号:wxid_rszr95aw79ie22)