艾滋病至今仍是全球重大公共卫生挑战,联合国艾滋病规划署数据显示,2024年全球约有3900万HIV感染者,年新增感染约130万例。
抗逆转录病毒治疗能有效抑制病毒复制,让感染者的预期寿命接近健康人群,却始终无法彻底清除体内的病毒,根治艾滋病的最大障碍,就是被称为“潜伏库”的病毒藏身之所——静息CD4+T细胞中处于休眠状态的HIV-1。
这些携带整合前病毒的静息 T 细胞在感染者体内广泛存在,可在实验室环境中,游离的HIV病毒颗粒却几乎无法感染静息T细胞,这一矛盾现象困扰了病毒学家和免疫学家数十年:为什么静息T细胞在体内容易被HIV攻陷,在体外却对病毒“油盐不进”?
近日,一篇发表在国际顶级期刊Nature上的研究论文,终于解开了这个困扰学界多年的谜题,而问题的核心答案,就藏在HIV最主要的传播方式之中。
这项由伦敦大学学院(现通讯作者单位为伦敦玛丽女王大学Blizard研究所)Clare Jolly教授团队领衔完成的研究,彻底推翻了“静息T细胞必须先经过全面激活才能被HIV感染”的传统认知,揭示了 HIV 利用细胞间接触主动改写宿主细胞防御规则的狡猾机制。

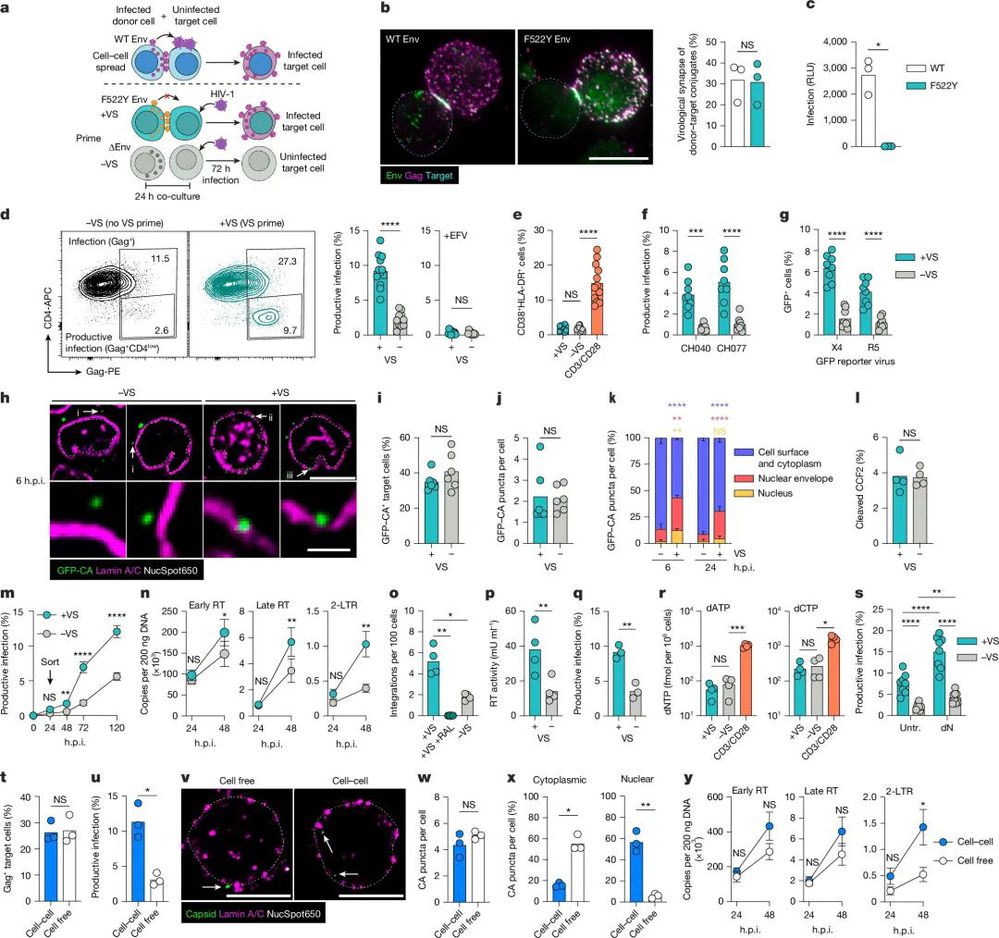
在此之前,学界早已明确HIV在体内主要通过两种途径感染细胞:一种是游离病毒颗粒的细胞感染,也是实验室最常用的感染方式;另一种是细胞间直接接触传播(CCS),也就是被感染的供体T细胞和未感染的靶细胞形成名为“病毒学突触(VS)”的紧密接触结构,实现病毒的高效传递。
过去学界普遍认为,细胞间传播的感染效率更高,只是因为突触结构能向靶细胞定向释放大量病毒,带来了更高的感染复数(MOI);而静息T细胞无法被游离病毒感染,是因为这类细胞处于非分裂的静息状态,缺乏病毒复制所需的关键条件,必须通过有丝分裂原激活,进入细胞周期后才能被HIV侵染。
甚至有观点认为,体内静息T细胞里的潜伏病毒,都来自那些曾经被激活、完成感染后又恢复静息状态的T细胞,静息T细胞本身无法被直接感染。
但这项新研究用严谨的实验设计,彻底颠覆了这些固有认知。研究团队首先设计了一套精妙的实验体系,把病毒学突触的形成、细胞间接触,和病毒的感染传递过程完全分离开来。
他们对HIV的包膜蛋白Env进行了单点突变(Env-F522Y),这种突变的Env依然能介导供体T细胞和靶静息T细胞形成完整的病毒学突触,却会让病毒失去膜融合和进入靶细胞的能力,无法完成感染。
研究人员用这种突变病毒感染供体T细胞,让其与自体静息CD4+T细胞共培养24小时完成“突触预刺激”,再移除供体细胞,用游离HIV去攻击这些经过预刺激的静息T细胞。
结果令人震撼:仅仅是一次没有发生病毒传递的细胞间接触,就让原本对游离病毒高度抵抗的静息T细胞,感染效率出现了极其显著的提升,且优先被感染的是静息记忆T细胞,这与HIV在人体内优先感染记忆T细胞的特征完全吻合。
病毒突触形成通过增强核输入驱动静息T细胞对HIV-1的易感性
更关键的是,研究团队通过超高分辨率即时结构照明显微镜(iSIM)、活细胞成像和分子生物学实验,精准锁定了静息T细胞感染的真正限速步骤——病毒衣壳的核输入,而非此前学界普遍认为的逆转录过程。
过去的研究认为,静息T细胞内的dNTP水平极低,加上SAMHD1蛋白对逆转录的限制,是病毒无法完成感染的核心原因。
但这项研究发现,突触预刺激完全没有改变静息T细胞内的dNTP水平,也没有影响SAMHD1的磷酸化状态和核糖核苷酸还原酶的表达;即便向细胞外添加外源性脱氧核苷,让逆转录的底物不再受限,也只能让所有组别的感染效率同步提升2倍,完全无法消除突触预刺激带来的感染优势。
与之相对的是,研究人员清晰地观察到,经过突触预刺激的静息 T 细胞中,6小时就有31%的病毒衣壳定位在核膜上、12%进入了细胞核;而未经刺激的对照组,仅有12%的衣壳到达核膜,进入细胞核的比例更是只有2%,且这种差距不会随着时间推移被弥补。
作为病毒核输入的替代标志物,2-LTR环状DNA的水平在预刺激组中出现了数十倍的提升,前病毒的整合效率也同步显著增加,直接证实了病毒衣壳的核输入,才是HIV攻破静息 T 细胞的最大瓶颈。
那么,细胞间的简单接触,究竟是如何打通这个核输入瓶颈的?
研究团队一步步拆解了这条由HIV操控的完整信号轴。当被感染的供体细胞通过Env与静息T细胞表面的CD4受体结合时,会立刻激活与CD4偶联的酪氨酸激酶LCK,启动下游的信号级联反应;这条信号通路会绕过经典的T细胞受体(TCR)激活通路,不会让静息T细胞表达CD69、CD25等活化标志物,也不会触发细胞进入分裂周期,却能特异性地激活周期蛋白依赖性激酶1(CDK1)。
CDK1本是调控细胞有丝分裂的核心激酶,主要在分裂期发挥作用,而研究人员发现,在静息T细胞中,CD4-LCK信号会通过抑制WEE1激酶、激活CDC25磷酸酶和CDK7,让CDK1发生激活型磷酸化,同时解除抑制型磷酸化,实现 CDK1 的短暂激活,整个过程完全不依赖细胞周期的推进。
被激活的CDK1,正是HIV打开细胞核大门的关键钥匙。细胞核与细胞质之间的物质交换,完全由核膜上的核孔复合体(NPC)管控,这个由三十余种核孔蛋白(Nups)组成的巨大复合体,是细胞内最精密的“分子安检门”,像HIV衣壳这样的超大尺寸货物,本极难穿过。
而CDK1会直接对核孔复合体上的核孔蛋白进行广泛的磷酸化修饰,尤其是组成核孔中央通道的Nup54、Nup62,以及核篮结构的核心蛋白TPR,引发核孔复合体的结构重塑。
研究人员通过定量质谱分析证实,CD4信号触发了核孔复合体胞质丝、中央通道、核篮三大结构域中多个核孔蛋白的磷酸化,而CDK1抑制剂能完全消除这种磷酸化修饰;超高分辨率显微成像也显示,经过Env刺激的细胞中,更多的HIV衣壳成功穿过核孔,定位到了核孔的核质侧,而对照组的衣壳大多停滞在核孔的胞质侧,无法完成跨核膜转运。
更有意思的是,这种核孔重塑并非只为HIV“开后门”,细胞自身的输入蛋白β1(KPNβ1)的核输入效率也同步提升,证实HIV通过细胞间接触,直接让整个核孔复合体进入了更宽松的“通行模式”。
这项研究还带来了一个颠覆性的范式更新:HIV的细胞间传播之所以在体内占据绝对主导地位,从来都不只是因为突触结构能传递更多病毒。研究团队证实,即便是已经被激活、本就对HIV易感的T细胞,细胞间接触触发的CD4-LCK-CDK1信号轴,依然能显著加速病毒衣壳的核输入,进一步提升感染效率。
这意味着,HIV在进化中形成了一套双重策略:通过细胞间接触,既实现了病毒的定向高效传递,又主动给靶细胞发放了感染“通行证”,双管齐下实现体内的快速扩散,完美解释了为什么在淋巴组织这种T细胞高度密集、细胞间接触频繁的环境中,HIV的传播速度会远超游离病毒感染的理论极限。
这项研究的意义,远不止解开了一个数十年的科学谜题。它彻底推翻了“静息T细胞必须预先全面活化才能被HIV感染”的经典教条,首次证实静息CD4+T细胞可以通过细胞间接触被HIV直接感染,清晰解释了体内HIV潜伏库的核心来源。
并非只有活化后恢复静息的T细胞会成为潜伏库,静息T细胞本身就能被直接感染并形成潜伏感染,这为我们理解HIV的体内潜伏机制,提供了全新的底层认知。
更重要的是,它为艾滋病的功能性治愈,开辟了全新的研发方向。潜伏库之所以难以清除,核心原因是我们既无法阻止新的潜伏库持续形成,也难以精准唤醒已经形成的潜伏病毒。
而这项研究发现的CD4-LCK-CDK1信号轴,正是阻止新潜伏库形成的绝佳靶点:如果能开发出靶向这条通路的抑制剂,就有望阻断HIV通过细胞间传播对静息T细胞的直接感染,从源头遏制潜伏库的补充。
同时,对这条信号通路的深入探索,也有望让我们找到可控激活静息T细胞核孔、反向唤醒潜伏病毒的方法,让“激活并杀死”的治愈策略,拥有更精准、更安全的实现路径。
当然,这项研究目前仍处于基础研究阶段,从机制发现到临床转化,还有很长的路要走。但它让我们再一次看清了这个古老病毒的狡猾与精妙——HIV甚至不需要进入细胞,仅仅通过细胞表面的受体结合,就能远程操控宿主细胞的核孔开关,为自己的感染铺平道路。
而对这条感染通路的破解,不仅让我们离艾滋病治愈的目标更近了一步,还为免疫学研究带来了意外的启发:CD4分子除了作为TCR的共受体参与抗原识别,还能通过独立的信号通路调控核孔复合体的功能,影响细胞的核质转运,这为我们理解CD4+T细胞的生理功能,打开了全新的窗口。
研究团队也指出,乙肝病毒、疱疹病毒等多种需要穿越核孔完成感染的病原体,同样依赖细胞间传播实现高效扩散,这种通过细胞接触调控核孔状态的机制,很可能是多种病毒通用的感染策略,未来的研究或将为更多传染病的防控,提供全新的思路。
[1]Mesner, D., Whelan, M.V.X., Shivkumar, M. et al. HIV-1 signalling remodels nuclear pores to licence infection. Nature (2026). doi:10.1038/s41586-026-10453-3