引言
免疫检查点分子是免疫稳态的关键调节因子,负责维持激活与耐受之间的平衡。在癌症中,肿瘤利用检查点通路抑制抗肿瘤免疫并促进进展。免疫检查点抑制剂(ICIs)的出现彻底改变了癌症治疗,特别是靶向临床验证的PDL1–PD1和CTLA4轴的药物。然而,大多数患者的获益有限或短暂,原因通常是检查点分子的失调。本文基于现有知识库,深入探讨在遗传、表观遗传、转录、转录后、翻译和翻译后水平上运作的多层次调控机制,这些机制共同塑造了肿瘤和免疫细胞中检查点的丰度和功能。
-02-
一、免疫检查点受体-配体相互作用及治疗性抗体
免疫系统依赖于精细调节的机制来对病原体和异常细胞产生有效反应,同时限制自身免疫和组织损伤。免疫检查点分子,即共刺激和共抑制配体-受体对,是这一调节的核心,它们校准免疫活动的幅度和持续时间。
在癌症中,这种平衡通过上调抑制性配体或受体和/或下调共刺激通路而向免疫逃逸倾斜。主要共刺激受体-配体对包括CD28-CD80/86、OX40-OX40L、4-1BB-4-1BBL等,而共抑制对则包括PD-1-PD-L1/2、CTLA-4-CD80/86、LAG3-MHC II、TIM3-CEACAM1、TIGIT-CD155等。
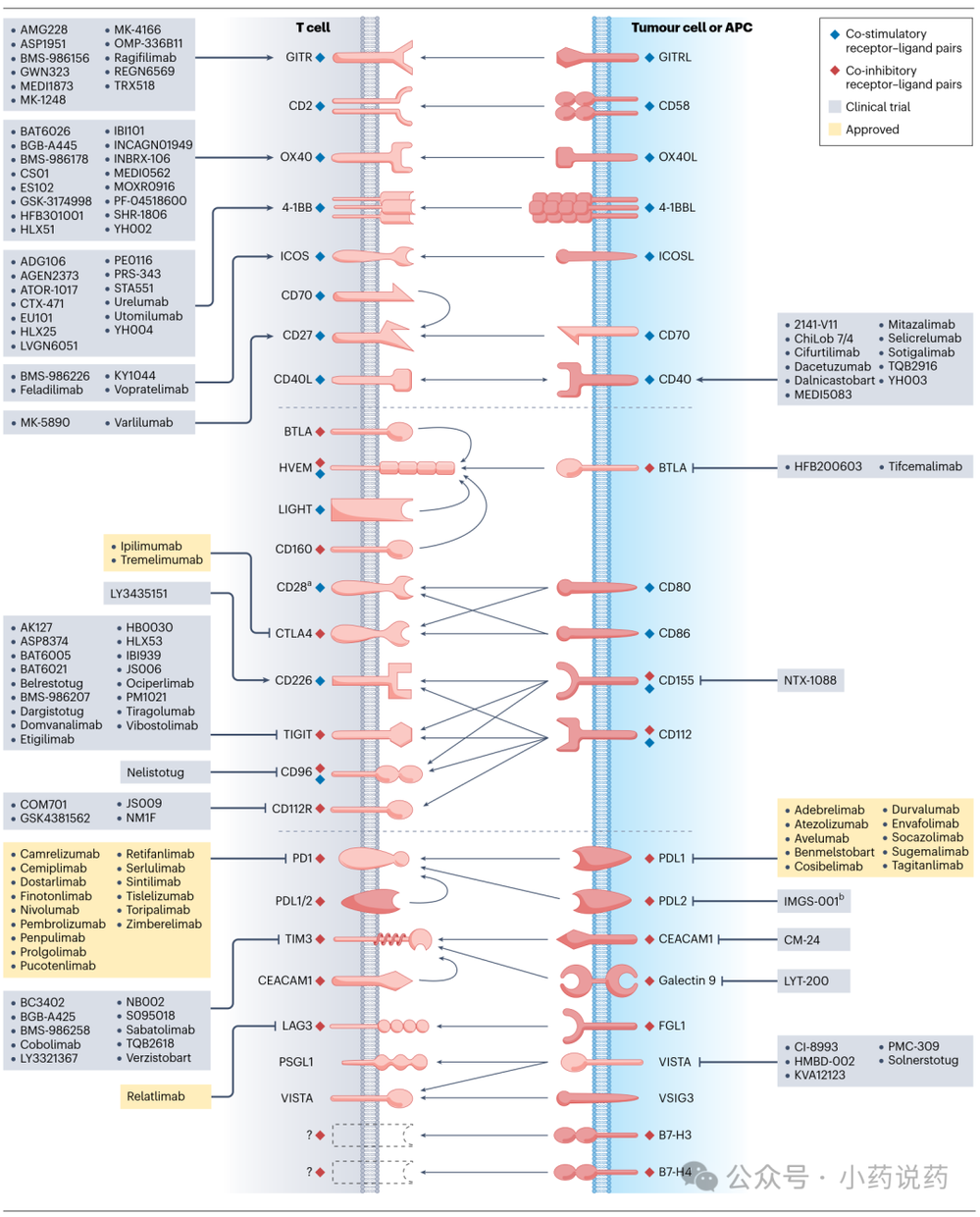
针对PD-1(如Pembrolizumab, Nivolumab)和PDL1(如Atezolizumab, Durvalumab)的抗体,以及针对CTLA-4(如Ipilimumab)的抗体,已在多种癌症治疗中获得批准。
抗PD-1和抗PDL1制剂主要在肿瘤微环境(TME)和淋巴组织中发挥作用,通过阻断PD1与肿瘤、髓系和其他基质细胞上的配体相互作用来重振耗竭的CD8⁺ T细胞。相反,抗CTLA4抗体主要通过阻断CTLA4与抗原呈递细胞上的CD80和CD86相互作用,并在一定程度上通过Fcγ受体介导的机制消耗肿瘤内调节性T细胞,从而增强T细胞激活。
-03-
二、免疫检查点基因的遗传变异
遗传变异通过多种直接机制影响免疫检查点生物学。
1. 单核苷酸变异(SNVs)与SNPs
顺式作用变异,包括启动子或增强子多态性、3'-非翻译区(UTR)突变和拷贝数变化,可改变基础转录本丰度或破坏转录后控制;结构变异可改变检查点基因完整性或去除调控元件,导致异常表达;影响检查点配体或受体的编码突变可直接影响癌症中的配体可用性和免疫参与。例如,CD274(编码PDL1)的3'-UTR突变破坏了其与通常抑制PDL1表达的microRNA miR-570的相互作用,导致PDL1上调。CTLA4种系变异rs231775导致CTLA4蛋白对APC上CD80的亲和力增加,从而增强T细胞抑制并升高肺、乳腺、食管和贲门癌的风险。
2. 结构变异与拷贝数变异
9p24.1扩增是检查点基因位点结构变异的一个突出例子,在霍奇金淋巴瘤和原发性纵隔大B细胞淋巴瘤(PMBCL)中复发,该区域包含CD274和PDCD1LG2(编码PDL2)基因,扩增导致其过表达。此外,破坏CD274 3'-UTR的结构变异消除了转录后抑制,导致T细胞白血病/淋巴瘤、弥漫性大B细胞淋巴瘤(DLBCL)和胃腺癌中的异常过表达。
3. 共刺激分子基因的缺失
在DLBCL中,CD58(也称为LFA3)和CD70的失活性突变或缺失是复发的,CD58缺失与CD19 CAR-T细胞疗法反应差相关。实验性CD58缺失赋予了对ICIs和CAR-T细胞或TCR-T细胞治疗的抗性。
-04-
三、免疫检查点的表观遗传调控
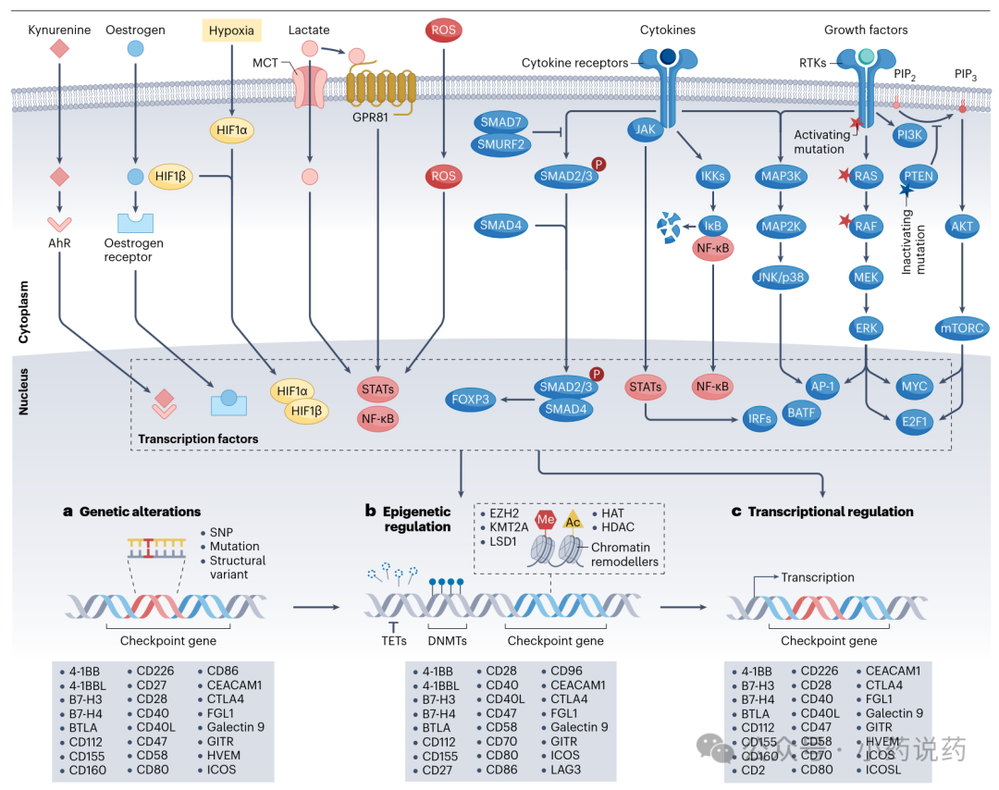
1. DNA甲基化
DNA甲基化通常在CpG二核苷酸的胞嘧啶上添加甲基,通常与转录沉默相关。在癌症基因组图谱(TCGA)队列中,共抑制基因的启动子倾向于低甲基化,而参与抗原呈递和共刺激的基因往往高甲基化。例如,IDH1或IDH2突变导致肿瘤代谢物D-2-羟戊二酸(D-2HG)积累,抑制TET双加氧酶,引发广泛高甲基化,包括检查点基因启动子,从而下调PDL1表达。
2. 组蛋白修饰
组蛋白修饰更易变,使基因能够快速、依赖环境地适应。
乙酰化:组蛋白乙酰转移酶(HATs)催化赖氨酸乙酰化,创造松散的染色质结构。例如,HAT1在胰腺癌中调节PDL1表达,敲低HAT1通过BRD4依赖机制降低PDL1表达。
甲基化:组蛋白甲基转移酶(HMTs)或去甲基化酶调节检查点表达。例如,赖氨酸甲基转移酶2A(KMT2A)介导CD274启动子的激活标记H3K4me3,驱动PDL1表达。LSD1通过去除激活的H3K4标记沉默Pdcd1;在慢性抗原暴露下,LSD1无法定位到Pdcd1位点,导致持续的PD1表达和T细胞耗竭。
3. 染色质重塑
SWI/SNF家族等染色质重塑复合物调节核小体位置。ARID1A(SWI/SNF复合物的核心亚基)失活性突变常见于卵巢和胃肠道癌,与PDL1表达增加相关。SMARCA4缺陷通过丧失SWI/SNF介导的检查点基因抑制,提高了基线和干扰素反应性PDL1表达。
四、免疫检查点的转录调控
1. 肿瘤内在机制
致癌信号通常汇聚于直接调节检查点基因的转录因子。MYC结合到CD274和CD47启动子,驱动PDL1和CD47表达。RTK–RAS–RAF–PI3K级联突变是研究最广泛的例子之一。例如,在ALK重排的非霍奇金淋巴瘤中,异常ALK信号增强STAT3与CD274启动子结合。在NSCLC中,EGFR突变激活ERK–JUN轴诱导PDL1。KRAS和BRAF突变通过JUN依赖性转录驱动NSCLC和CRC中的PDL1。
2. 环境刺激——细胞因子
免疫抑制性细胞因子:TGFβ和IL-10是检查点介导抑制的关键协调者。在卵巢癌中,IL-10诱导巨噬细胞中的B7-H4表达,并驱动CD8⁺ T细胞上的LAG3和PD1上调。TGFβ通过BATF促进HAVCR2, LAG3, TIGIT和CTLA4的转录。
促炎细胞因子:IFNγ是PDL1和PDL2的特征性诱导剂,通过JAK-STAT1-IRF1轴作为经典的适应性免疫抵抗机制起作用。IFNγ还上调CD47,帮助逃避免疫吞噬。TNF主要通过NF-κB信号诱导前列腺癌和结肠癌细胞系中的PDL1。
3. 环境刺激——代谢物和物理因素
乳酸激活NF-κB,诱导肝细胞癌模型中中性粒细胞的PDL1转录。活性氧(ROS)通过NF-κB驱动PDL1表达。犬尿氨酸激活T细胞中的芳香烃受体,诱导PD1、CTLA4和LAG3的表达。缺氧主要通过稳定缺氧诱导因子1α(HIF1α)诱导PDL1表达。
五、免疫检查点的转录后调控
转录后过程,包括可变剪接、mRNA稳定性控制、RNA修饰和非编码RNA相互作用,精细调节蛋白输出。
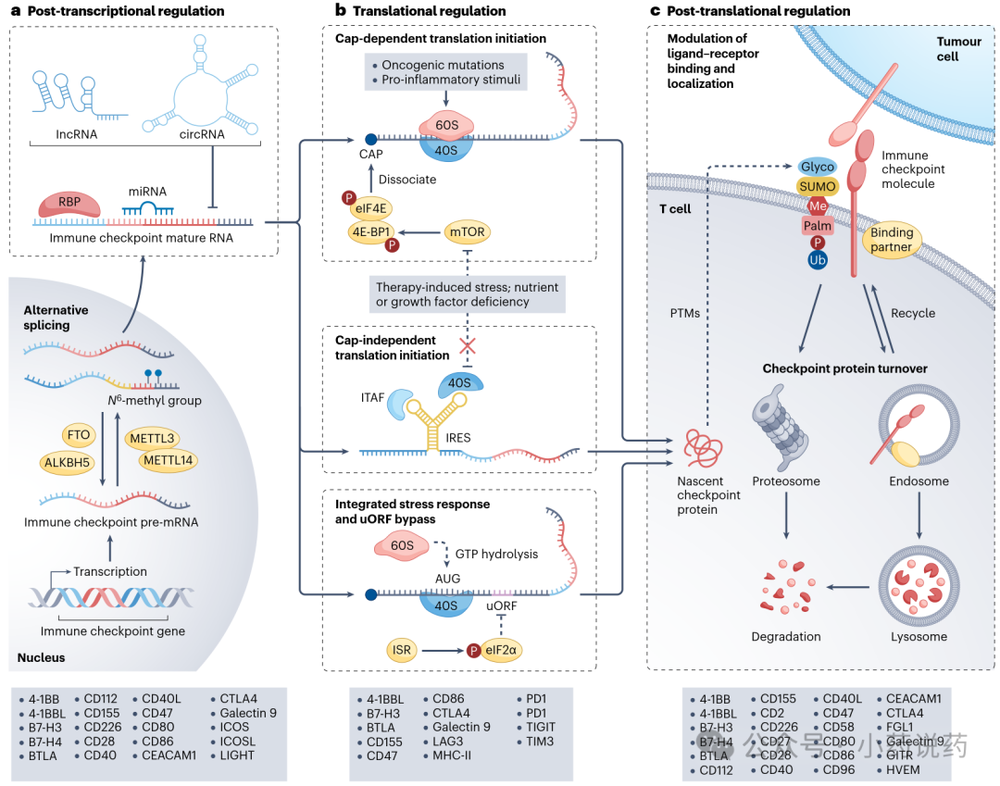
1. 可变剪接
可变剪接产生功能不同的变体。CD274前mRNA可剪接产生缺乏跨膜结构域的可溶性PDL1(sPDL1)异构体,这些异构体作为诱饵结合T细胞上的PD1或隔离治疗性抗PDL1抗体。临床上升高的循环sPDL1与ICI反应差相关。CTLA4前mRNA可剪接产生可溶性CTLA4,由人黑色素瘤细胞系分泌。
2. mRNA稳定性控制
RNA结合蛋白调节检查点mRNA稳定性。例如,Tristetraprolin(TTP)结合CD274 3'UTR中的AU富含元件并招募衰变机制。ELAVL1(HuR)通过稳定CMTM6 mRNA间接保护PDL1免受溶酶体降解。
3. RNA修饰
N⁶-甲基腺苷(m⁶A)是mRNA内部最常见的修饰。在膀胱癌细胞中,JNK信号上调METTL3,增加CD274 3'UTR中的m⁶A沉积,IGF2BP1结合这些修饰转录本,稳定CD274 mRNA。YTHDF1促进CRC中甲基化抑制性检查点转录本的翻译。
4. 非编码RNA
miRNAs:miR-200家族和miR-34a/c直接靶向CD274,减少PDL1水平。
lncRNAs:MALAT1结合并灭活miR-195和miR-17-5p,解除对PDL1和B7-H4的抑制。NEAT1在CD8⁺ T细胞中吸附miR-155,导致TIM3表达增加。
circRNAs:hsa-circ-002178吸附miR-34a,阻止PDL1抑制。
六、免疫检查点的翻译调控
1. 帽依赖性和非依赖性翻译
癌症细胞经常增强帽依赖性翻译。eIF4E过度激活促进黑色素瘤细胞中的PDL1表达和卵巢癌细胞中的B7-H3表达。在结直肠癌细胞系中,mTOR抑制剂治疗阻断帽依赖性翻译,但矛盾地通过CD274 5'-UTR中的IRES增加PDL1合成。
2. 应激下的翻译
整合应激反应(ISR)激活导致eIF2α磷酸化和帽依赖性翻译的抑制,但允许核糖体绕过抑制性uORF,在主要ORF处起始,从而增强特定转录本的翻译。ISR驱动的eIF2α磷酸化缓解了uORF介导的PDL1和CD155抑制。
3. 应激颗粒
在TCR刺激后,人外周血T细胞中形成的应激颗粒暂时隔离未翻译的mRNA,选择性存储编码抑制性受体的转录本,延迟其翻译并延长效应器活性。
七、免疫检查点的翻译后调控
1. 检查点蛋白稳定性
PDL1经历泛素化和蛋白酶体降解。CUL3–SPOP E3连接酶泛素化PDL1,加速其周转。去泛素化酶CSN5在TNF信号期间拯救PDL1免于降解。N-糖基化保护PDL1免受泛素化和蛋白酶体降解。棕榈酰化通过膜锚定和防止溶酶体降解来稳定检查点蛋白,PDL1在Cys272处被ZDHHC9和ZDHHC3棕榈酰化。
2. 亚细胞运输和定位
PDL1与CMTM6在质膜和回收内体中结合,保护表面PDL1免受溶酶体和蛋白酶体降解,同时使其回收至细胞表面。乙酰化限制PDL1的核进入,而HDAC2介导的去乙酰化解除了这一限制,PDL1入核后可作为转录调节因子。SUMO化导致CD155在细胞内被隔离。
3. 检查点配体-受体相互作用
N-连接糖基化,特别是polyLacNAc链添加,是高效PDL1–PD1结合所必需的。PDL1可与CD80形成顺式异源二聚体,隔离PDL1使其不与PD1结合,并保护CD80免受CTLA4介导的内吞,从而保留CD80–CD28共刺激信号。
八、精准患者分层的新兴生物标志物
在经典霍奇金淋巴瘤中,涉及CD274和PDCD1LG2的9p24.1改变高达97%的病例,驱动PDL1和PDL2过表达,并与PD1阻断的敏感性密切相关。相反,功能丧失突变可作为阴性预测因子,如T细胞恶性肿瘤中的PDCD1突变。
2. 表观遗传谱
检查点启动子低甲基化使抑制分子能够组成性过表达。KMT2家族组蛋白甲基转移酶突变的患者在多个ICI治疗队列中显示出更好的结果。
3. 基因表达模式和微环境预测因子
肿瘤中的IFNγ相关mRNA谱预测对PD1阻断的临床反应。耗竭T细胞的转录程序,特别是由PD1水平和TIM3、LAG3、TIGIT协调上调定义的独特耗竭状态,具有预测潜力。
九、靶向检查点调控
靶向控制检查点表达的调控机制,而非检查点蛋白本身,提供了调节抗肿瘤免疫的新机会。
1. 破坏肿瘤内在的抑制性检查点表达
针对致癌驱动因子(如KRAS(G12C))的抑制剂与PD1阻断联合,在肺癌和结肠癌临床前模型中显示出协同抗肿瘤作用。BET抑制是一种有前景的方法,BRD4整合致癌转录程序与检查点控制,药理学BET抑制不仅抑制肿瘤细胞增殖信号,还降低PDL1水平。
2. 抑制TME诱导的抑制性检查点表达
在晚期肿瘤中,TGFβ信号驱动细胞毒性淋巴细胞排斥。在CRC小鼠模型中,TGFβ通路抑制与PD1或PDL1阻断联合,协同增加肿瘤内T细胞浸润。PGE₂驱动TME介导的免疫抑制,靶向胞外ATP-P2RY2轴已被证明可与ICI、CAR-T细胞、TCR-T细胞和TIL疗法协同作用。
3. 恢复共刺激检查点表达
增强或恢复内源性受体-配体表达比使用激动剂抗体可能更安全、更生理。DNMT抑制剂可逆转DNA甲基化,重新激活沉默的共刺激分子,例如卵巢癌细胞中的CD70重表达。HDAC抑制剂可解除免疫刺激程序的抑制,在骨髓瘤患者的骨髓培养中,HDAC抑制剂处理上调了肿瘤细胞和树突状细胞上的CD80, CD86, MHC-I和MHC-II。
结语
免疫检查点不是简单的开关,而是跨解剖部位、细胞类型和分子层运作的相互连接的调控网络中的动态节点。这种系统层面的观点有助于解释为什么阻断单个检查点通常只会产生短暂的益处:肿瘤通常在基线时参与多种部分冗余的逃逸机制,并在治疗压力下主动重新连接这些回路以恢复免疫抑制。其他治疗方式,包括化疗、放疗和靶向治疗,也会以可能无意中促进免疫逃逸的方式扰乱检查点调节。因此,从ICI中获得持久益处需要更深入地了解检查点通路在个体患者中何时、何地以及如何重新连接。
参考资料:

以上直播课和PPT均可在知识星球中免费下载,扫描下方二维码加入“小药说药知识文库”的知识星球

公众号已建立“小药说药专业交流群”微信行业交流群以及读者交流群,扫描下方小编二维码加入,入行业群请主动告知姓名、工作单位和职务。
