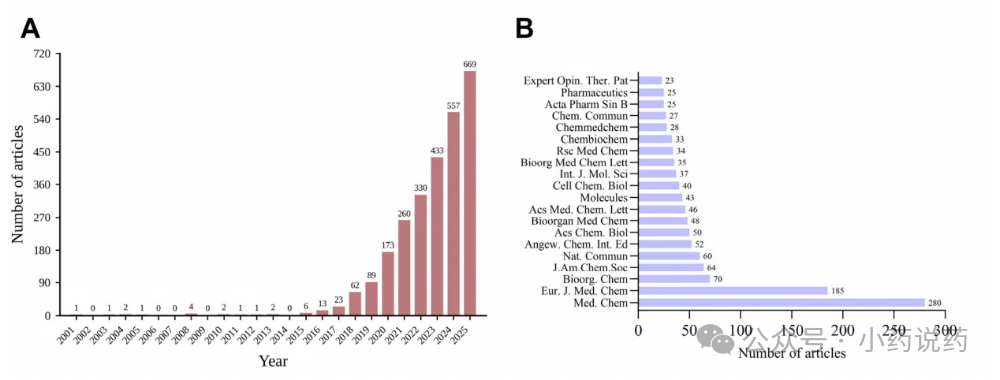
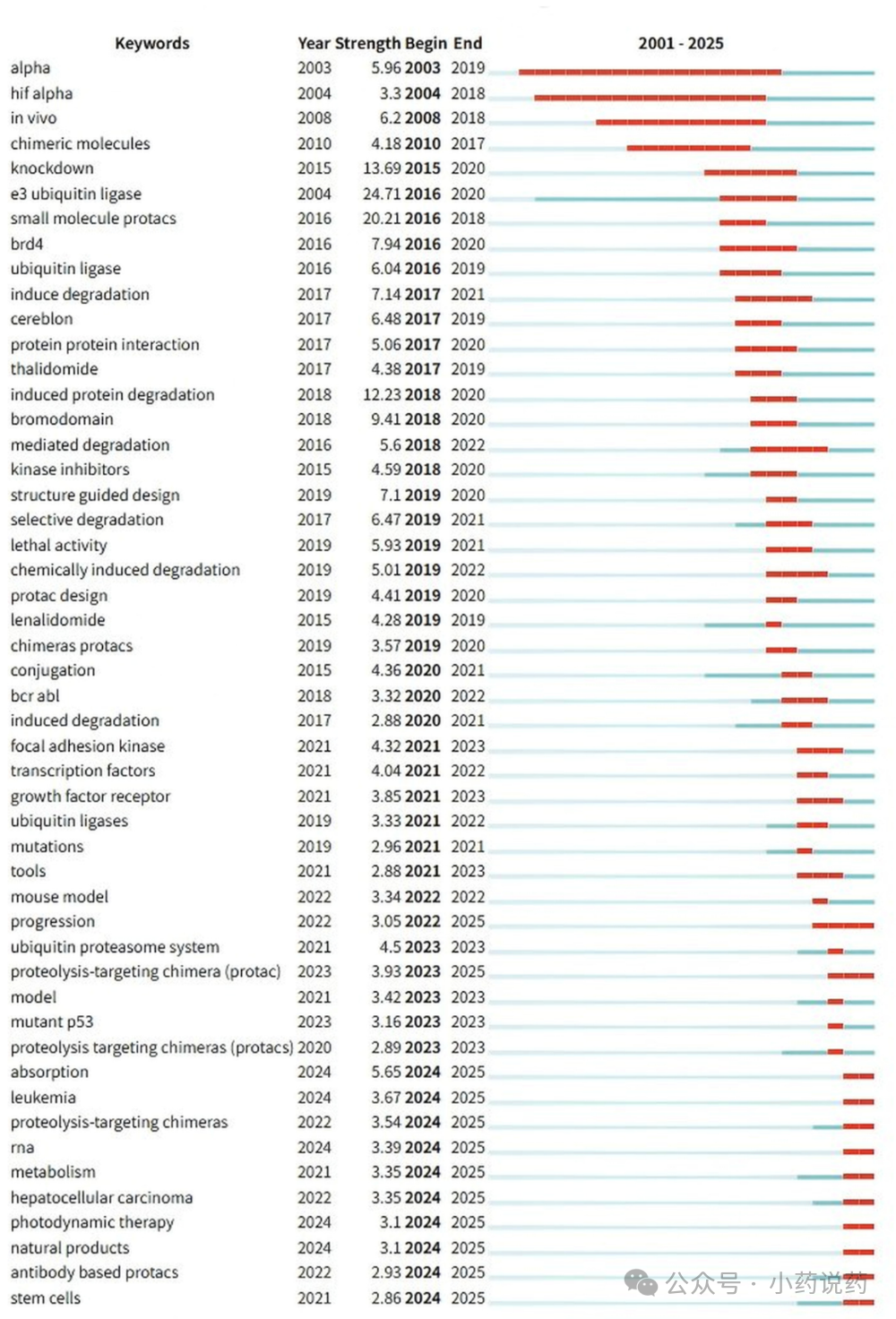
三、五大新兴前沿:下一代PROTACs将走向何方?基于对共被引文献和关键词的时间线分析,该研究凝练出了当前最值得关注的五大新兴研究方向,它们将共同定义PROTACs的未来。1. 从“亲和力”到“三元复合物”:理性设计的新基石在最新的研究阶段(2021-2025),“结构基础”成为了最大的关键词集群之一。这标志着理解上的深刻转变:决定PROTAC效力和选择性的,已不再是其对靶蛋白或E3连接酶的单体亲和力,而是其诱导形成的“靶蛋白-PROTAC-E3连接酶”三元复合物的稳定性与协同性。这一发现催生了全新的设计策略:通过连接子刚性化或大环化来预先组织PROTAC的生物活性构象,从而降低熵罚,提升降解活性和选择性。同时,为了摆脱对CRBN和VHL这两种常用E3连接酶的过度依赖,拓展E3连接酶工具箱(如DCAF16、RNF114、KEAP1等)成为了克服耐药和实现组织特异性降解的关键。2. 攻克耐药性:EGFR与KRAS的正面战场面对耐药突变,传统激酶抑制剂往往束手无策,而PROTACs因其降解机制,有望成为破解这一难题的“银弹”。这催生了以“EGFR”为核心的抗耐药研究集群。例如,针对第三代EGFR抑制剂奥希替尼耐药的C797S三突变,已有多个PROTAC分子(如HJM-561)展现出强大的体内外降解活性和口服生物利用度。在KRAS这个“不可成药”的经典靶点上,PROTACs同样取得了突破,不仅有针对G12C突变体的LC-2,更有能覆盖13种临床相关突变体的pan-KRAS降解剂出现,多个分子已进入临床I期。3. 突破“五规则”:口服生物利用度的攻坚战PROTACs的分子量通常在800-1100 Da之间,远超传统小分子药物的“类药五规则”(Ro5),这使其口服吸收和生物利用度成为最大挑战之一。如今,“吸收”一词的强势爆发,正说明了这一难题正从“认知”走向“解决”。核心策略是“分子变色龙”:这些大分子在极性水溶液中舒展,暴露氢键供体;但在穿越亲脂的细胞膜时,它们通过分子内氢键折叠成紧凑的构象,屏蔽极性表面。通过精妙的连接子工程和立体化学优化,已有多个PROTAC(如ARV-766、ACBI2)实现了临床级别的口服药代动力学,证明这一难题是“可解的”。4. 进军中枢神经系统与肿瘤免疫:应用场景的扩展PROTACs的舞台已不再局限于肿瘤学。神经退行性疾病:作为全新的研究热点,“tau”、“α-突触核蛋白”和“神经退行性疾病”作为独立集群出现。针对tau蛋白和α-突触核蛋白的PROTACs已在动物模型中展现出改善认知功能的潜力,首个靶向LRRK2的PROTAC(ARV-102)已进入临床I期。其核心优势在于:即便因血脑屏障限制导致药物浓度很低,其催化降解模式仍能发挥药效。精准递送:为了解决PROTACs的“脱靶毒性”,一系列精准递送和条件激活策略应运而生,如抗体-PROTAC偶联物(DACs)、核酸适配体-PROTAC偶联物(APCs),以及由光、缺氧等肿瘤微环境信号激活的前药策略和纳米递送系统,旨在将降解活性精准地限制在病灶部位。5. 计算驱动:AI与MD模拟的加速器最后一个新兴前沿是“分子动力学(MD)模拟”集群的出现。面对PROTACs巨大的化学空间(连接子、靶蛋白配体、E3配体三者的排列组合),传统的实验筛选效率低下。如今,计算化学和人工智能正从“辅助解读”走向“前瞻设计”。全原子MD模拟已被用于预测三元复合物的构象和稳定性,并指导连接子设计。同时,机器学习模型也在尝试直接预测PROTACs的降解活性(DC50)和细胞通透性。尽管数据量和模型精度仍有待提升,但其与实验反馈的结合,有望实现PROTACs的自动化闭环设计。 总结从2001年的概念诞生,到2025年的III期临床成功,PROTACs用了二十五年完成了从“奇思妙想”到“临床现实”的跨越。本研究的计量学分析清晰地表明,这个领域正从基础科学问题(“能不能降解”)全面转向转化医学问题(“如何制成好药”)。口服生物利用度的优化、E3连接酶库的拓展、CNS疾病的渗透、精准递送策略的开发以及AI驱动的理性设计,构成了PROTACs未来发展的核心驱动力。我们或许正站在下一代革命性疗法的门口。 参考文献:Sakamoto, K.M., Kim, K.B., Kumagai, A., Mercurio, F., Crews, C.M., & Deshaies, R.J. (2001). Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences, 98(15), 8554-8559.Bondeson, D.P., Mares, A., Smith, I.E.D., et al. (2015). Catalytic in vivo protein knockdown by small-molecule PROTACs. Nature Chemical Biology, 11, 611-617.Zengerle, M., Chan, K.-H., & Ciulli, A. (2015). Selective Small Molecule-Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chemical Biology, 10(8), 1770-1777.Lu, J., et al. (2015). Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chemistry & Biology, 22(6), 755-763.Gadd, M.S., Testa, A., Lucas, X., et al. (2017). Structural basis of PROTAC cooperative recognition for selective protein degradation. Nature Chemical Biology, 13(5), 514-521.Bondeson, D.P., Smith, B.E., Burslem, G.M., et al. (2018). Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chemical Biology, 25(1), 78-87.Lai, A.C. & Crews, C.M. (2017). Induced protein degradation: an emerging drug discovery paradigm. Nature Reviews Drug Discovery, 16(2), 101-114.Raina, K., Lu, J., Qian, Y., et al. (2016). PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proceedings of the National Academy of Sciences, 113(26), 7124-7129.Schapira, M., Calabrese, M.F., Bullock, A.N., & Crews, C.M. (2019). Targeted protein degradation: expanding the toolbox. Nature Reviews Drug Discovery, 18(12), 949-963.Bekes, M., Langley, D.R., & Crews, C.M. (2022). PROTAC targeted protein degraders: the past is prologue. Nature Reviews Drug Discovery, 21(3), 181-200.