二甲双胍想必大家都已经很熟悉了,作为2型糖尿病治疗领域当之无愧的「常青树」,始终稳坐降糖药界的C位。
一直以来,学界普遍认为,二甲双胍的降糖作用主要来自肝脏。其经典机制是抑制肝脏糖异生,也就是减少肝脏自行制造葡萄糖,从而帮助控制血糖水平。
然而,事实上,常规口服二甲双胍后,肠道内的药物浓度可达到血浆的300倍,以及肝脏的10-100倍。而且在糖耐量异常和新发2型糖尿病患者中,二甲双胍不仅没有降低内源性肝糖生成,甚至还升高了肝糖生成。
这就耐人寻味了。如果抑制肝脏产糖是二甲双胍发挥降糖作用的核心机制,那为什么药物会大量富集于肠道?又为何在肝糖生成并未明显下降,甚至有所升高的情况下,依然能够稳定发挥降糖作用?
难不成,二甲双胍降糖的关键,不在肝脏?
近期,来自美国西北大学的研究团队发表于Nature Metabolism的一项研究就狠狠挑战了“二甲双胍的降糖作用主要来自肝脏”的经典理论。这项研究发现,二甲双胍能够特异性抑制肠道上皮细胞中的线粒体复合体Ⅰ,将肠道转变为一个持续消耗葡萄糖的「代谢池」,从而显著降低餐后血糖水平。

DOI:10.1038/s42255-026-01530-y
研究人员首先分析了大量服用二甲双胍的人群数据,结果发现,在所有代谢物中,瓜氨酸的水平下降得最明显。而肠道是人体唯一能够在外周合成瓜氨酸的器官,并且瓜氨酸的合成过程高度依赖线粒体提供的ATP。
也就是说,二甲双胍很可能通过损害小肠线粒体的功能,使线粒体“无力”合成瓜氨酸,从而显著降低了血液中瓜氨酸水平。
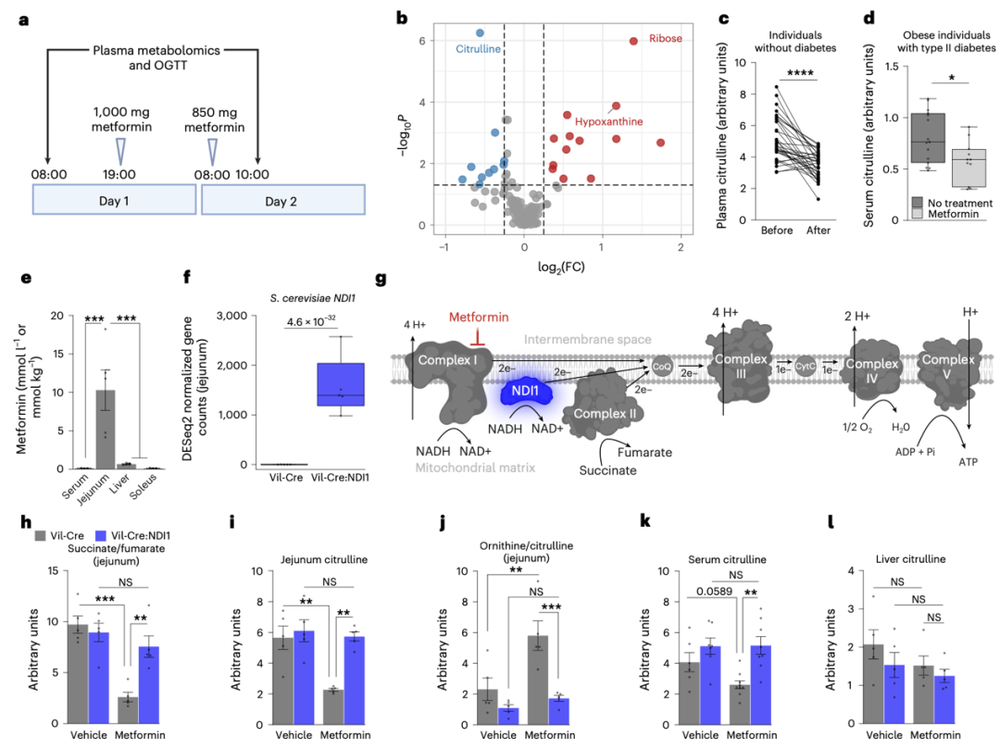
二甲双胍通过抑制肠道上皮线粒体复合体I,下调肠道瓜氨酸合成,降低机体循环瓜氨酸水平
顺着这条线索,研究人员精准锁定了线粒体复合体I。他们在雄性小鼠的肠道上皮细胞中转入了一个来自酵母的“外挂”基因NDI1(Vil‑Cre:NDI1小鼠)。这种基因编码的蛋白,相当于给线粒体呼吸链装了一条“备用通道”。
如果说复合体I是“正门”,NDI1就是旁边的“侧门”。正常情况下,葡萄糖要氧化供能,电子必须通过复合体I这扇“正门”。二甲双胍的作用,恰恰就是死死堵住这扇“正门”。 但那些经过改造的Vil‑Cre:NDI1小鼠(肠道上皮细胞特异性表达NDI1),即便正门被堵死,电子依然能通过NDI1这一“侧门”悄悄溜进去,让线粒体维持基本运转。
研究团队给普通雄性小鼠喂二甲双胍后,它们肠道中的瓜氨酸水平明显下降。同时,肠道对葡萄糖的摄取量却急剧飙升。但给Vil‑Cre:NDI1雄性小鼠喂同等剂量的二甲双胍,它们瓜氨酸水平却没有明显变化,肠道也不怎么“抢”葡萄糖了。
这也意味着,二甲双胍发挥降糖作用,靠的是抑制肠道上皮细胞的线粒体复合体I。
既然锁定了靶点,那二甲双胍具体是怎么降血糖的呢?
研究人员进一步分析后发现,二甲双胍抑制了肠道上皮细胞的线粒体复合体I之后,这些细胞立刻陷入了“能量危机”。为了自救,它们开始疯狂进行糖酵解。
通俗点说,原本肠道上皮细胞是“细水长流”地慢慢利用葡萄糖来发电。可线粒体复合体I被“堵住”之后,它们就不再精打细算了,转而开始“胡吃海塞式”地消耗葡萄糖。
结果,肠道变成了一块巨大的“葡萄糖海绵”。餐后血糖升高时,大量的葡萄糖还没来得及进入血液去“祸害”血管和器官,半路就被肠道这块海绵直接吸走了。吸进去的葡萄糖被迅速转化成乳酸和乳酰-苯丙氨酸(Lac-Phe)这种代谢物。
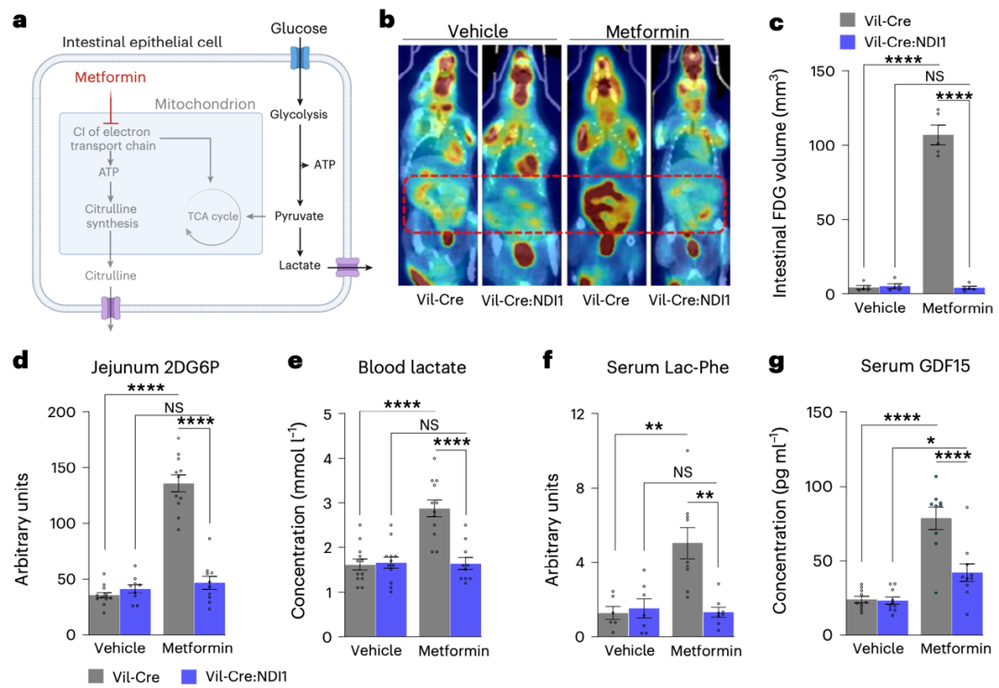
二甲双胍通过抑制小肠上皮线粒体复合体I,促使肠道大量摄取循环葡萄糖、亢进糖酵解,进而升高血乳酸和Lac-Phe水平
事实上,Lac-Phe本身就是一种天然的食欲抑制剂。它的升高,同样依赖于肠道线粒体复合体I被抑制。二甲双胍这一招堪称釜底抽薪,既拦住了餐后血糖的“洪峰”,又抑制了食欲,简直是一箭双雕。
总体而言,上述研究揭示了,二甲双胍降糖的关键并非传统认为的抑制肝脏产糖,而是通过特异性抑制肠道上皮细胞中的线粒体复合体I,使肠道陷入“能量危机”并激活糖酵解,将其转化为一块巨大的“葡萄糖海绵”,在餐后迅速吸走大量葡萄糖,转化为乳酸和具有食欲抑制作用的乳酰-苯丙氨酸(Lac-Phe),从而既降低了餐后血糖,又帮助控制体重。
[1]Sebo, Z.L., Chakrabarty, R.P., Grant, R.A. et al. Metformin inhibits mitochondrial complex I in intestinal epithelium to promote glycaemic control. Nat Metab (2026). https://doi.org/10.1038/s42255-026-01530-y