引言
乳腺癌是全球女性最常见的恶性肿瘤之一,发病率逐年上升。乳腺癌的治疗格局已深刻进入精准分子分型和个体化治疗的时代。尽管取得了进展,但内在和获得性耐药、显著的肿瘤异质性和靶点逃逸仍是实现持久临床缓解的主要瓶颈。
抗体药物偶联物(ADCs)是由单克隆抗体通过化学连接子与细胞毒性药物(称为载荷)连接而成的免疫偶联物。ADC是一种基于载体的化疗,可选择性地将强效细胞毒性药物递送至肿瘤内部。目前,ADCs是癌症治疗的重要类别,迄今为止美国食品药品监督管理局已批准了十七种ADCs。尽管癌症治疗取得了所有进展,但内在和获得性耐药仍然是成功治疗的主要障碍。
解决上述挑战的一个前瞻性方法是将连接子-载荷复合物与双特异性抗体(BsAbs)偶联,从而产生了双特异性抗体药物偶联物(BsADCs)的概念。这种双靶向方法的临床原理在HER2阳性乳腺癌中曲妥珠单抗和帕妥珠单抗联合治疗的成功中得到了有力证明。通过分别靶向HER2的不同结构域,这种标准治疗方案可协同抑制HER2信号传导,并已成为转移性和早期HER2阳性乳腺癌的标准治疗,提供了强有力的概念验证。与传统ADCs相比,BsADCs独特的双表位/靶点结合模式不仅能够结合实体瘤中共表达的抗原以增强选择性,还能显著改善内化。这些独特优势使BsADCs成为下一代ADCs领域的重要力量
-02-
一、BsADC的结构
BsADC的核心结构包括双特异性抗体、连接子和载荷。在与癌细胞表面抗原结合后,BsADC通过受体介导的内吞作用被内化,载荷通过连接子或抗体在内溶酶体区室中的降解而释放。因此,BsADCs的开发需要仔细考虑多个因素,包括靶抗原生物学、抗体特异性、载荷细胞毒性和作用机制、连接子稳定性和裂解性以及偶联位点。
除了这些因素外,双特异性靶向的一个固有考虑是,为了获得最佳活性,可能需要在同一肿瘤细胞上共表达两种靶抗原。这一概念理论上可能限制与单特异性ADC相比的靶细胞群体。然而,这种潜在限制被两个关键因素所抵消。首先,对双阳性细胞的增强内化和效力可能更有效地根除这一关键亚群。其次,也是至关重要的一点,使用可裂解连接子能够实现“旁观者效应”,即在双阳性细胞中释放的膜渗透性载荷可以扩散并杀死仅表达一种甚至不表达靶抗原的邻近肿瘤细胞。该效应对于克服肿瘤异质性和确保全面的肿瘤细胞杀伤至关重要。
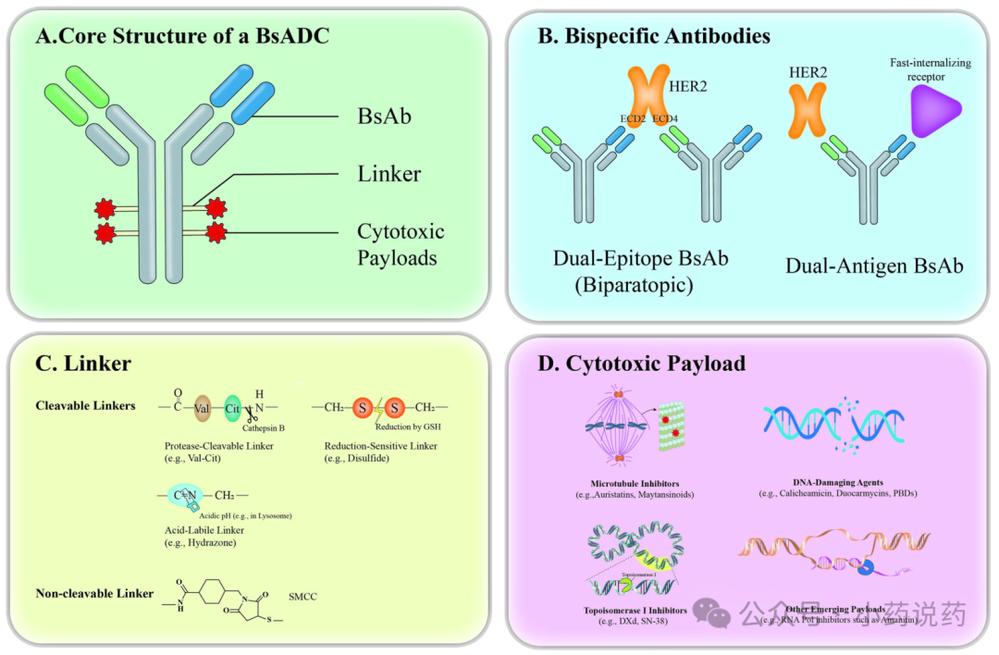
BsADCs的结构整合了多个关键设计组件,这些组件共同决定了其治疗功效和安全性。如上图所述,这些包括双特异性抗体格式、连接子化学和细胞毒性载荷,每种都提供了独特的战略优势。
-03-
二、靶向HER2的BsADCs
乳腺癌是全球女性最常见的癌症类型,其中HER2阳性乳腺癌占所有乳腺癌病例的15%至20%。临床上批准的HER2靶向单克隆抗体和ADCs已显著改变了HER2阳性乳腺癌的治疗选择,带来了临床获益。然而,HER2的表达异质性限制了当前抗HER2疗法在HER2水平相对较低的情况下的疗效。这不仅缩小了HER2治疗的适应症范围,而且在治疗压力下也助长了耐药性。此外,HER2对内化的潜在抵抗降低了药物疗效。幸运的是,BsADCs的双重结合模式为当前乳腺癌HER2靶向治疗中内化不良和耐药性的挑战提供了一个有前景的解决方案。
靶向HER2的双表位BsADC
ZW49:ZW49是通过蛋白酶可裂解连接子将新型N-酰基磺酰胺类奥瑞他汀毒素载荷偶联到双特异性抗HER2 IgG1抗体ZW25(现通用名zanidatamab)的链间二硫键半胱氨酸上生成的。基于奥瑞他汀毒素的载荷,如MMAE,是强效的微管抑制剂,对多种癌细胞系的IC50值通常在10至100 pM之间。这种新型变体中的理化修饰旨在微调其膜渗透性,从而控制旁观者效应并扩大治疗窗。
Zanidatamab是一种使用Azymetric平台生产的不对称IgG1样分子,该平台结合了工程化的CH3结构域(“杵臼”和静电引导)以确保重链异源二聚化和正确组装。这种靶向铰链区半胱氨酸残基的定点偶联策略能够生成具有可控DAR为2-3的高度均质的ADC群体,如疏水相互作用色谱所表征,最大限度地减少了随机偶联常见的异质性和聚集风险。该偶联物在循环中表现出高稳定性。
临床前数据表明,ZW49表现出强效的肿瘤杀伤效果和良好的耐受性,且不影响HER2亲和力(最高非严重毒性剂量=18 mg/kg)。目前,ZW49正在进行I期临床试验。初步结果表明,其在各种类型的HER2阳性肿瘤中具有前景的疗效。在队列扩展推荐剂量下治疗的八名乳腺癌患者(中位既往治疗线数为六线)中,观察到ORR为13%(8名患者中有1名)。毒性分析显示两例持续超过14天的G2角膜炎;约43%的患者出现角膜炎,但所有事件均降至G1或最终消退。未发生间质性肺病事件或与治疗相关的死亡。然而,ZW49随后的临床开发似乎被Zymeworks管线内的其他资产降级,其未来的开发策略仍有待澄清。
MEDI4276:MEDI4276是通过将曲妥珠单抗的单链可变片段连接到抗HER2全人源mAb 39S(IgG1κ)重链的N末端构建的。曲妥珠单抗和39S结合HER2上非重叠的远距离表位。因此,MEDI4276包含四个抗原结合位点,每条臂上两个,能够与HER2相互作用。四个高亲和力结合位点的存在促进了HER2分子的有效募集和聚集,即使在低HER2表达的肿瘤模型中也是如此。
载荷AZ13599185是阿斯利康/MedImmune开发的微管蛋白抑制剂tubulysin的变体。Tubulysins是一类超强效细胞毒素,通常表现出低纳摩尔到皮摩尔的IC50值。双表位抗体在Fc区包含三个位点突变:L234F、S239C和S442C。每条重链上的两个工程化半胱氨酸残基(S239C和S442C)通过马来酰亚胺己酰基连接子实现AZ13599185与抗体的定点偶联,从而产生DAR为4的双表位ADC。L234F突变与S239C突变结合降低了FcγR结合。认为这些突变可能最大限度地减少FcγR介导的、HER2非依赖性ADC被正常组织摄取,从而降低脱靶毒性。
一项首次人体、I期、多中心、开放标签、剂量递增研究评估了MEDI4276在HER2阳性晚期乳腺癌或胃癌患者中的安全性、药代动力学、免疫原性和抗肿瘤活性。该研究表明,尽管在乳腺癌中表现出一些临床活性(尽管有限),但MEDI4276的进一步临床开发受到不利的PK特征(不足以克服潜在的抗原沉没)和高毒性的挑战。MEDI4276剂量从0.05 mg/kg递增至0.9 mg/kg(每3周60-至90分钟静脉输注)。在0.9 mg/kg时超过最大耐受剂量,两名患者出现剂量限制性毒性:3级肝功能检测值升高,其中一名还伴有3级腹泻,后缓解。另外两名患者在较低剂量(0.4和0.6 mg/kg)时报告了3级肝功能检测值升高的DLTs。在意向治疗人群中,有一例完全缓解(0.5 mg/kg;乳腺癌)和两例部分缓解(0.6和0.75 mg/kg;乳腺癌)——所有患者既往均接受过曲妥珠单抗、帕妥珠单抗和恩美曲妥珠单抗治疗。MEDI4276具有可证明的临床活性,但在剂量>0.3 mg/kg时显示出不可耐受的毒性。
JSKN003:JSKN003是一种双特异性HER2靶向ADC,它利用了KN026的双表位结合。Fc聚糖定向偶联平台允许每个抗体定点附着四个DXd分子,与基于马来酰亚胺的随机半胱氨酸偶联相比,增强了结构均一性。这种方法还改善了亲水性,减少了聚集,并增强了血清稳定性,从而延长了载荷保留时间。这些特性有助于形成有利的药代动力学特征并减少脱靶毒性,预计这将扩大治疗窗。
JSKN003进入了一项首次人体、剂量递增、多中心、开放标签的I期研究。针对具有不同HER2表达水平的各种实体瘤的32名患者的I期临床试验取得了有希望的结果。治疗耐受性良好,3级或以上TRAEs发生率为6.3%(2/32),表现为贫血(血液学)和疲劳(体质性)。最常见的任何级别TRAEs是腹泻(62.5%)和恶心(50.0%)。重要的是,未观察到剂量限制性毒性,且在剂量高达8.4 mg/kg时未达到最大耐受剂量。JSKN003观察到的显著更高的DCR(90.6%)可能表明其具有持续疾病控制的潜力。基于这些令人鼓舞的初步疗效和安全性信号,JSKN003于2023年12月进入III期试验。
靶向HER2的双抗原BsADC
同时靶向HER2和快速周转受体(如CD63、PRLR和APLP2)的双靶向BsADCs可以进一步优化HER2靶向ADCs的内化和溶酶体运输,有可能增强细胞毒性并降低HER2的临床应用阈值。值得注意的是,这些创新策略虽然在临床前概念验证中表现出说服力,但尚未进入临床试验,凸显了将这些设计转化为治疗药物的挑战。
BsADC on HER2×CD63:CD63是四次跨膜蛋白超家族的成员,表达广泛但并非普遍。它主要分布在细胞表面、晚期内体和溶酶体。CD63的胞质结构域包含一个YXXO共有基序,使其能够与衔接蛋白相互作用并募集网格蛋白包被。这种与AP-2/AP-3和网格蛋白的相互作用使CD63成为一种衔接蛋白,促进相关蛋白从细胞表面到晚期内体-溶酶体区室的扩展运输。CD63在这些细胞区室中的存在使其成为旨在增强内化和溶酶体运输的BsADCs的潜在靶点,最终改善药物递送和治疗效果。
BsADC on HER2×PRLR:催乳素受体是一种I型细胞因子受体,在一部分乳腺癌中表达,并可能有助于其发病机制。它在约25%的人类乳腺肿瘤中相对过表达,而在一些正常人类组织(包括乳腺)中表达水平较低。与HER2相比,PRLR被快速且持续地内化,并有效地转运到溶酶体进行降解。PRLR胞质结构域促进其自身快速的持续性内化和溶酶体降解。关键的是,当HER2通过双特异性抗体在细胞表面与PRLR交联时,这一特性被利用来将HER2共同纳入相同的快速内化和降解途径。这种强制伙伴关系显著增强了HER2的降解,从而改善了偶联的细胞毒性载荷的递送和疗效。低水平的细胞表面PRLR足以介导PRLR ADCs的有效杀伤。
BsADC on HER2×APLP2:APLP2是淀粉样前体蛋白家族中普遍表达的成员。APLP2胞质尾中包含的酪氨酸含有重叠的基于酪氨酸的NPXY和YXXØ基序。类似地,APLP2可以与AP-2结合,介导有效的内化,并在网格蛋白介导的内吞作用后引导至溶酶体降解。
BsADCs on HER2和HER3
人表皮生长因子受体家族成员是正常细胞生长和发育的有效介质。ErbB家族由四个密切相关的I型跨膜酪氨酸激酶受体组成:EGFR、HER2、HER3和HER4。受体二聚化(异源或同源二聚化)是ErbB功能和这些受体信号传导活性的基本要求。HER2-HER3异源二聚体被认为是最有效的ErbB对,具有相互作用强度、配体诱导的酪氨酸磷酸化和下游信号传导,并作为致癌单位发挥作用。
研究人员构建了一种HER2/HER3靶向的双特异性ADC,并表征了其理化性质、靶点特异性和体外内化,并评估了其在乳腺癌细胞系和动物模型中的抗肿瘤活性。HER2/HER3靶向的BsADC的DAR为2.89,表现出强效和选择性的抗肿瘤活性。
-04-
三、靶向EGFR × HER3的BsADC
EGFR(也称为ERBB1或HER1)是ERBB受体酪氨酸激酶家族的成员,该家族还包括HER2、HER3和HER4。EGFR在调节上皮恶性肿瘤的基本功能中起关键作用,从而诱导体内稳态紊乱。然而,由于治疗压力诱导的获得性基因组改变,靶向EGFR的单克隆抗体和TKIs通常导致临床耐药性的出现。BsADCs有望解决抗EGFR耐药机制,包括致敏突变和旁路通路的激活。
HER3由ERBB3基因编码。HER3本身没有激酶活性,但它能够与HER2(和/或EGFR)形成异源二聚体,这显著增加了反式磷酸化和下游信号级联的激活。对EGFR靶向治疗的耐药也可能通过ERBB受体的代偿性信号传导发生,涉及HER2和HER3等受体,这表明了联合治疗的潜力。然而,临床数据表明,帕妥珠单抗和西妥昔单抗的组合导致重叠毒性,使得该组合无法耐受。HER3在EGFR耐药的癌细胞系中显著过表达,促进了对EGFR的内在和获得性耐药以及PI3K的激活。
通过利用抗EGFR Fab和抗HER3 scFv,构建了靶向EGFR和HER3的BsAb,命名为SI-B001。拓扑异构酶I抑制剂ED04通过新型可裂解AC连接子偶联到SI-B001上的半胱氨酸位点,生成了一种新型BsADC,命名为BL-B01D1,报告的平均DAR约为8。ED04属于拓扑异构酶I抑制剂类别,以其高效力而闻名。BL-B01D1实现了对EGFR依赖性肿瘤的靶向杀伤,并减弱了HER3诱导的耐药性。
最近的临床数据为其疗效和安全性特征提供了强有力的证据。在一项I期研究中,入组的局部晚期或转移性HER2-(IHC 0、1+或2+/ISH-)乳腺癌患者接受BL-B01D1治疗,剂量为2.5 mg/kg,每三周一次(D1D8 Q3W)。入组患者未根据EGFR或HER3表达水平进行筛选。截至2024年9月30日,在121名可评估患者(中位既往治疗线数=3)中,客观缓解率为42.1%,确认的ORR为36.4%,疾病控制率为80.2%。中位无进展生存期为6.9个月,中位缓解持续时间为9.7个月。令人鼓舞的是,在包括所有HER2表达亚组中均观察到疗效。
尽管疗效令人鼓舞,但EGFR×HER3靶向药物(如BL-B01D1)的安全性特征需要仔细考虑。I期临床数据显示,最常见的治疗相关不良事件主要是血液学毒性(例如中性粒细胞减少症、白细胞减少症、血小板减少症)和胃肠道毒性(例如恶心、口腔炎、呕吐),这些是拓扑异构酶I抑制剂基ADCs的典型表现,并且可以通过支持性护理进行管理。3级及以上TRAEs主要是血液学毒性,能够通过标准支持措施(包括剂量减少)有效管理,如TRAE导致的停药率为5.0%所证明。重要的是,未显著报告心脏毒性或细胞因子释放综合征的信号,也未观察到ILD事件。这一临床特征表明,虽然理论风险存在,但BL-B01D1的实际毒性可能主要由强效载荷驱动,并且可以在临床上进行管理。持续保持警惕对于全面表征其长期安全性仍然至关重要。
-05-
四、靶向TROP2 × HER3的BsADC
Trop2是一种I型表面糖蛋白,在胚胎器官发育中至关重要,其在正常组织中的表达有限。越来越多的证据表明,Trop2在一系列实体瘤中过表达,显著影响肿瘤生长、侵袭和转移。因此,Trop2已成为实体瘤有吸引力的预后标志物和治疗靶点。首个Trop2靶向抗体药物偶联物Trodelvy的批准,提高了转移性乳腺癌和转移性尿路上皮癌的生存率,这鼓励了对Trop2生物学和扩展Trop2靶向治疗策略的进一步研究。TROP2和HER3在三阴性乳腺癌中均高表达,并与较差的生存率相关,为双靶向提供了强有力的原理基础。
JSKN016是一种双特异性TROP2/HER3靶向ADC,采用新型连接子与载荷偶联,DAR为4。虽然具体化合物未公开,但ADCs中使用的拓扑异构酶I抑制剂(例如DXd、SN-38)普遍以其高效力和跨细胞膜扩散的能力为特征。这种极端细胞毒性和膜渗透性的结合是观察到的mTNBC队列中显著初步疗效和潜在旁观者效应的根本驱动因素。
正在进行的首次人体I期研究的临床数据已开始显现出其有前景的特征。截至2024年12月23日,在一组经过大量预处理的转移性三阴性乳腺癌患者中,JSKN016显示出显著的抗肿瘤活性。在跨多个剂量水平(4–8 mg/kg Q3W)入组的5名可评估疗效的mTNBC患者中,ORR为80.0%(4/5名患者达到部分缓解),其余患者达到疾病稳定并伴有29.5%的肿瘤缩小。在报告时尚未达到最大耐受剂量,PFS数据尚未成熟。尽管抗肿瘤活性引人注目,但这种TROP2×HER3双特异性ADC的安全性特征需要持续评估。
JSKN016-101试验的初步数据揭示了一个独特的安全性特征:最常见的治疗相关不良事件是恶心、呕吐和口腔粘膜炎。在8 mg/kg剂量水平发生了一例3级痤疮样皮炎的剂量限制性毒性。≥3级TRAEs不常见,仅在10.5%的患者中观察到,包括中性粒细胞减少症、淋巴细胞减少症和口腔粘膜炎。至关重要的是,未观察到心脏毒性、细胞因子释放综合征或治疗相关ILD的信号,并且没有不良事件导致治疗中断或死亡,支持JSKN016在研究剂量范围内的临床耐受性。
-06-
结语
总之,双特异性抗体药物偶联物代表了乳腺癌治疗中一个充满希望且快速发展的前沿领域,具有重塑治疗范式的潜力。然而,通往临床成功的道路充满了相互关联的挑战。首先,临床毒性特征是被转化而非消除。其次,双特异性格式的复杂工程和严格制造造成了生产瓶颈和高成本,直接影响患者可及性。最后,基本的科学障碍仍然存在,包括管理免疫原性、克服肿瘤抗原异质性以及确保连接子稳定性以防止脱靶毒性。因此,BsADCs的未来取决于协同努力应对这一三重挑战。成功将需要靶点、连接子和生物标志物的持续生物学优化,与制造创新和对临床安全性特征的清晰理解无缝整合。只有通过这种整体方法,才能实现更有效和可耐受的个体化治疗。
参考文献:
Recent advances in bispecific antibody-drug conjugates for breast cancer therapy. Cancer Chemother Pharmacol. 2026 Jan 24;96(1):15.
公众号已建立“小药说药专业交流群”微信行业交流群以及读者交流群,扫描下方小编二维码加入,入行业群请主动告知姓名、工作单位和职务。
