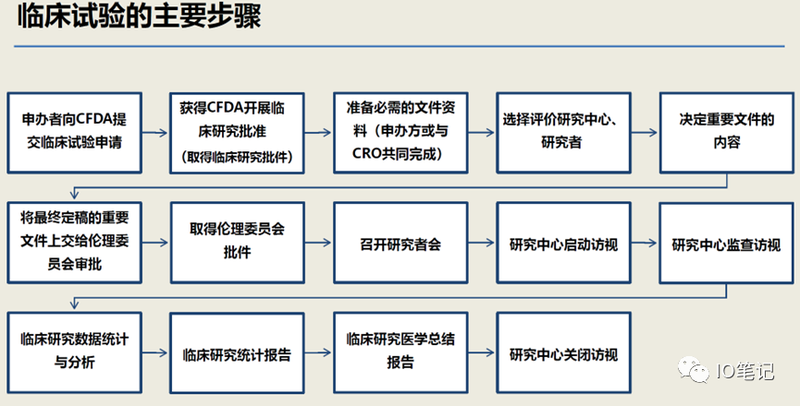
备注:图片来源于网络,侵删!
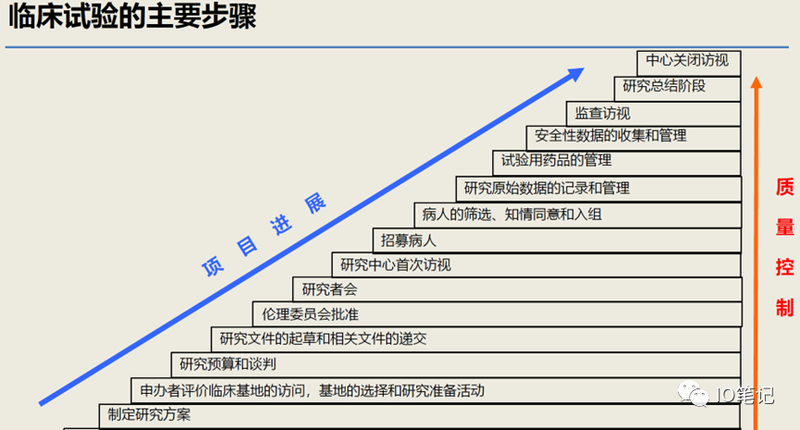
图片来源于网络,侵删!
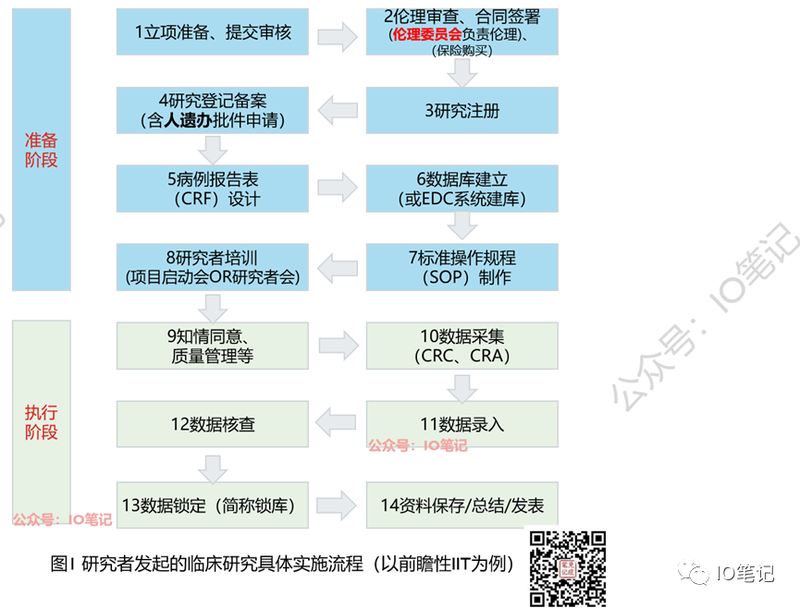
研究者发起的临床研究步骤:
临床试验区分;

备注:图片来源于网络,侵删!
常见专业术语如下:
01 临床研究机构:医院都有专门的网址介绍;
例如华西医院的:http://www.cd120.com/clinicalresearch.html

伦理委员会(EC):英文全称Ethics Committee
由医学专业人员、法律专家及非医务人员组成的独立组织,其职责为核查临床试验方案及附件是否合乎道德,并为之提供公众保证,确保受试者的安全、健康和权益受到保护。
该委员会的组成和一切活动不应受临床试验组织和实施者的干扰或影响。
伦理委员会的存在主要是确保临床试验以受试者的利益为先。伦理委员会类似于站在受试者的立场上的专业的陪审团。
物临床试验质量管理规范(GCP)英文全称Good Clinical Practice
中文名称为"药物临床试验质量管理规范"。药品临床试验管理规范(GCP)是临床试验全过程的标准规定,制定GCP的目的在于保证临床试验过程的规范,结果科学可靠,保护受试者的权益并保障其安全。
研究者:英文全称Investigator
实施临床试验并对临床试验的质量及受试者安全和权益的负责者。研究者必须经过资格审查,具有临床试验的专业特长、资格和能力。
每一项临床试验有一位研究者总负责,其他人员在其指导和协调之下进行工作。这一位研究者称为主要研究者或研究者组长(PrincipalInvestigator,PI),他的主要助手称为合作研究者(Co-Investigator),其他参加的人员则称为协助研究者(Sub-Investigator,Sub-I)(临床研究的负责人)。
助理研究者(Sub-I):英文全称sub-investigator,
其他参加人员,如研究助理、研究护士等。
协调研究者(COI):英文全称Coordinating Investigator,COI,在多中心临床试验中负责协调参加各中心研究者工作的一名研究者。
主要研究者(PI):英文全称Principal Investigator,PI。
根据ICH-GCP的定义,Principal Investigator同Investigator实际上是一回事。如果一个药物临床研究机构(国外称为Site)只有一个研究者,就称为Investigator,如果一个机构有多位研究者,那么主要负责的研究者就称为PI,其他称为Sub-investigator(辅助研究者),常常简写为Sub-I。PI或者Sub-I的定义,在国外非常清晰。
02申办方
申办者(Sponsor)英文全称Sponsor:
发起一项临床试验,并对该试验的启动、管理、财务和监查负责的公司、机构或组织。申办方是“大Boss”。一项新药的研发及新药的临床试验都由大Boss组织。并且,最重要的是,大老板是负责发工资的。
03协助方
合同研究组织(Contract Research Organization, CRO)
指通过签订合同授权,执行申办者或者研究者在临床试验中的某些职责和任务的单位。合作研究组织,一种商业性公司或机构。申办方可委托其执行临床试验中的某些工作和任务。新药研发合同外包服务机构 。主要通过合同形式向制药企业提供新药临床研究服务的专业公司。
通过签订合同授权,执行申办者或者研究者在临床试验中的某些职责
CRO主要是协助申办者,派遣临床研究监察员(CRA)的公司。
可理解为CRO是“大Boss”的助理,帮助大老板处理某些业务。

区别:

现场管理组织(Site Management Organization,SMO)
直接协助研究者并提供Study Coordinator的服务,履行研究者授予的职责。
SMO主要协助研究者,是派遣CRC的公司。
申办方或CRO委托SMO进行临床研究,签订临床研究合同,CRO将临床试验的费用支付给SMO。SMO再与其紧密联系的各家研究者签署合同,根据研究者在临床研究中的实际工作量,付给研究者报酬。
临床监查员(CRA)英文全称Clinical Research Assistant,中文称临床监查员
临床监查员是申办者与研究者之间的主要联系人。CRA对外要管理site,对内要管理项目组的方方面面,协调和处理矛盾能力要求高。
其人数及访视的次数取决于临床试验的复杂程度和参与试验的医疗机构的数目。临床监查员应有适当的医学、药学或相关专业学历,并经过必要的训练,熟悉药品管理有关法规,熟悉有关试验药物的临床前和临床方面的信息以及临床试验方案及其相关的文件。其监查的目的是为了保证临床试验中受试者的权益受到保障,试验记录与报告的数据准确、完整无误,保证试验遵循已批准的方案和有关法规。
临床研究协调者(CRC)英文全称Clinical Research Coordinator,临床研究协调者。
负责临床试验的落实工作;负责跟踪研究进度及临床试验工作协调,重要的一项是筛选入组患者的数据收集、录入EDC系统等。
主要职责如下:
□与伦理委员会和申办者、CRA之间的联络;
□协助研究者实施试验的各项工作,如
· 获取知情同意书;数据收集与CRF转录‘
□应对监管机构、申办者和CRA的监查、稽查与检查;
□... ...
临床试验中的其他术语:
项目经理(Project Management,PM):临床研究项目管理,从本质上说,就是一个项目管理,跟工程项目、金融项目等一样,需要总体协调大家的配合和前进,做好时间、质量等几方面的总控。需要较强的综合能力,沟通、管理、团队合作和领导力、多任务协同,更重要的一点是,需要有医药相关背景。一般是有大局观、对各个角色都有所了解、资深的CRA等的转岗过来的。
物警戒(PV):
随着国内法规对于药物安全的要求越来越高,这个职位主要是跟进一些不良反应数据采集或者报送,对药物警戒部门的一些文件进行维护,定期更新标准操作流程,需要对药事法规和药物警戒条例比较了解,以保证不良反应的上报是合规的。
临床试验相关专业术语:
知情同意书(ICF)英文全称Informed Consent Form,ICF,
是每位受试者表示自愿参加某一试验的文件证明。研究者必须向受试者说明试验性质、试验目的、可能的受益和危险、可供选用的其他治疗方法以及符合《赫尔辛基宣言》规定的受试者的权利和义务者,使受试者充分了解后表达其同意。
病例报告表(CRF):英文全称Case Report Form,病例报告表,
是在临床试验中用以记录每一名受试者在试验过程中的症状、体征或实验室检查数据的文件。
不良事件(AE)英文全称Adverse Event, AE,
指病人或临床试验受试者接受一种药品后出现的不良医学事件,但并不一定与治疗有因果关系。
严重不良事件(SAE):英文全称Serious Adverse Event,
链接:不良反应(AE)评定的国际标准(CTCAE)详解(笔记43)
临床试验过程中发生的需住院治疗、延长住院时间、伤残、影响工作能力、危及生命或死亡、导致先天畸形等事件。
盲法/设盲
英文全称Blinding/masking,系指按试验方案的规定,在试验结束之前不让参与研究的受试者或研究者,或其他有关工作人员知道受试者被分配在何组(试验组或对照组),接受的是何种处理尤其是监视员在盲法试验中必须自始至终地保持盲态,从而避免他们对试验结果的造成人为干扰。
试验用药品
英文全称Investigational Product,用于临床试验中的试验药物、对照药品或安慰剂。
试验方案(Protocol):英文全称Protocol,
叙述试验的背景、理论基础和目的,试验设计、方法和组织,包括统计学考虑、试验执行和完成的条件。方案必须由参加试验的主要研究者、研究机构和申办者签章并注明日期。
研究者手册(IB):英文全称Investigator‘s Brochure,是有关试验药物在进行人体研究时已有的临床与非临床研究资料。
药品上市许可持有人(MAH):英文全称Marketing Authorization Holder,MAH,药品上市许可持有人。通常指拥有药品技术的药品研发机构、科研人员、药品生产企业等主体,通过提出药品上市许可申请并获得药品上市许可批件,并对药品质量在其整个生命周期内承担主要责任的制度。
英文全称World Health Organization,WHO,是联合国下属的一个专门机构,总部设置在瑞士日内瓦,只有主权国家才能参加,是国际上最大的政府间卫生组织。
药品使用:
bid 一日两次
tid 一日三次
qd 每天一次
临床试验其他专业术语;
CDE Center for drug evaluation 国家药品监督管理局药品审评中心
IIST industry-sponsored clinical trial 医药企业发起的临床试验
IIT Investigator-Initiated Clinical Trial 研究者发起的临床试验
CRP Clinical Research Physician 临床研究医生
FIH First-in-Human 首次人体试验
CO Clinical Operation 临床运营
DM Data Management 数据管理
EDC Electronic Data Capture 电子数据采集系统
SSU study start up 研究启动前准备工作
SEV site evaluation visit 中心评估访视
PSV pre-study visit 试验前访视
SIV site Initiation visit 中心启动访视
RMV routine monitoring visit 常规监查访视
COV/SOV close out visit/site close-outvisit 中心关闭访视
IB Investigator’s Brochure 研究者手册
IP Investigational Product 研究产品
ICF Informed Consent Form 知情同意书
SAP statistical analysis plan 统计分析计划
PD protocol deviation 方案偏离
PV protocol violation 方案违背
SAE serious adverse event 严重不良事件
SUSAR suspicious unexpected serious adversereaction 可疑的非预期的严重不良反应
CM Concomitant Medications 合并用药
MH medical history 既往病史
SPL Study Personnel List 研究人员名单
SSL Subject Screening Log 受试者筛选表
SEL Subject Enrollment Log 受试者入选表
BMI body mass index 体重指数
DBL Data base Lock 锁定数据库
IP investigational product 研究药品
FAS Full analysis set 全分析集
PPS per protocol set 符合方案集
SS safety set 安全分析集
CSR clinical study report 临床研究报告
FR Final Report 总结报告
QA Quality Assurance 质量保证
QC Quality Control 质量控制
BE bioequivalence 生物等效性
NDA 申报注册(欧洲称为MAA)
IND 申报临床
DLT Dose-restricted toxicity /Dose limiting toxicity 剂量限制性毒性
MTD maximal tolerated dose 最大耐受剂量
SCV siteclos-out visit 研究结束访视
备注:整合来自网络的相关公众号内容;真没有查到原始的出处~~