作者|小麦
近些年来,随着技术的进步,双抗药物得到快速发展。2024年,全球双抗药物市场规模达到约130亿美元。
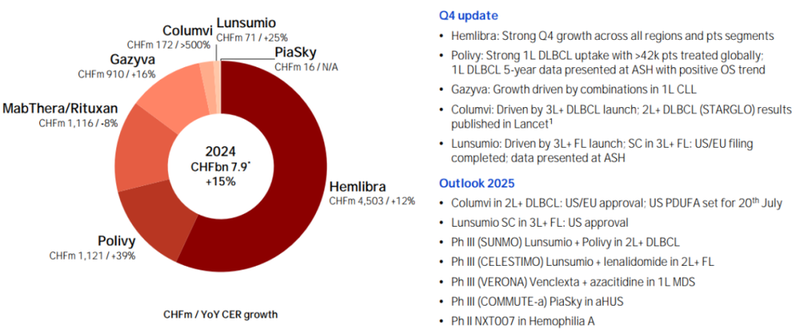
其中罗氏的用于治疗血友病的Hemlibra全球销量最高,2024年销售额达到45.03亿瑞士法郎(约53.61亿美元)。其次是罗氏的眼科药物Vabysmo增长迅速,2024年销售额为38.64亿瑞士法郎(约46.0亿美元),加上Columvi和Lunsumio,罗氏双抗规模已超百亿美元,占有绝对领先地位(图1)。
在我国,康方生物的两款双抗药物(开坦尼和依达方)表现突出,2024年合计销售额预计超过3亿美元。这表明中国药企在全球双抗市场中已占据重要地位。

01
新颖双抗结构和技术
早在1960年,纽约罗斯威尔公园纪念研究所的Nisonoff及其合作者在Science发表的论文中首次提出了双特异性抗体的概念。但是直到 2009年4月,首个双抗Catumaxomab卡妥索单抗被欧洲药品管理局(EMA)批准上市。截止目前,全球已获批17款双抗药物(图1)。
从获批的双抗药物结构中,我们可以看到双抗技术得到了快速发展,双抗的设计和生产逐渐迎来了革新。
早期的双特异性抗体主要通过化学偶联或杂交杂交瘤技术制备,但存在制备复杂、稳定性差等问题。
后来随着基因工程、杂交瘤技术、生物大分子重组等技术的飞速发展,双抗的技术和类型都得到很大的改善,新的双抗技术脱颖而出,包括BiTEs、DART和TandAb等。
BiTE免疫疗法(Bispecific T-cell Engager,双特异性T细胞衔接器)是一种创新的免疫治疗技术,即以T细胞作为效应细胞的双特异性单链抗体,它是采用单链抗体技术,将两个不同抗体的重链及轻链可变区连接在一条多肽链上。
BiTEs具有两个抗原结合部位,可以同时和T细胞及癌细胞表面的抗原分子结合,从而有效地激活效应T细胞来达到杀伤病变细胞的目的(图2)[1]。
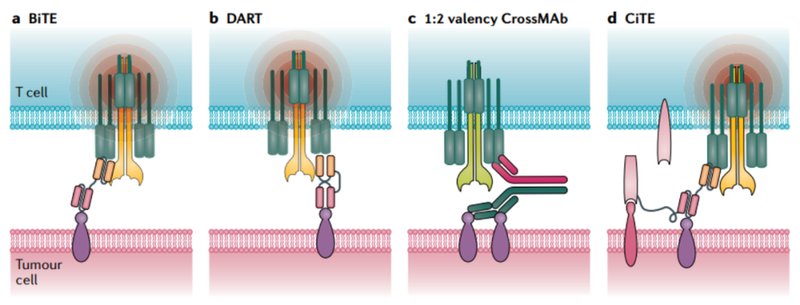
图2. BiTE和DART示意图
BiTEs具有以下优点:
1.高效性,能够显著增强T细胞对肿瘤细胞的杀伤作用,即使是在T细胞数量较少的情况下。
2.特异性,通过靶向肿瘤特异性抗原,BiTE疗法能够精准地攻击肿瘤细胞,减少对正常细胞的损伤。
3.广泛适用性,BiTE疗法可以针对多种肿瘤特异性抗原进行设计,具有广泛的应用潜力。
DART技术(Dual-Affinity Re-Targeting)是由MacroGenics公司和Servier公司联合开发的一种双特异性抗体构建技术。
与BiTEs相比,DART具有更有利的结构和生物学特性,包括更大的稳定性和将T细胞毒性靶向恶性细胞的最佳重定向。
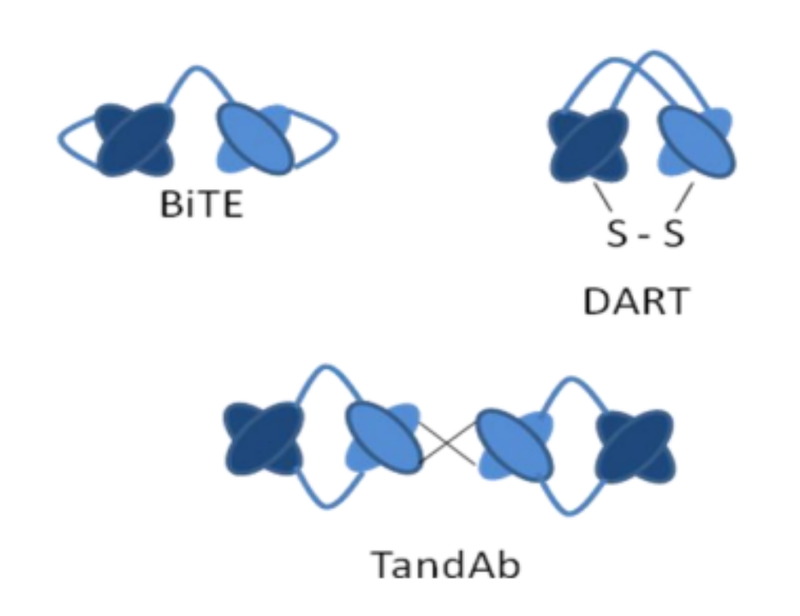
TandAb是一种四价双特异性抗体,由两条多肽链反向配对形成同源二聚体分子,其结构为Fv1-Fv2-Fv2-Fv1,包含4个单链可变片段(scFv),能够同时结合两种抗原,且每种抗原有两个结合位点,这种结构显著增强了抗体的结合能力和效力(图3)。
TandAb可以通过多种方法生成,包括重组DNA技术、化学交联和融合蛋白方法。其中,重组DNA技术是主要的生成方式,通过将两个不同的Fv片段串联,形成四价双特异性分子。
TandAb技术凭借其独特的结构设计和高效的功能,在双特异性抗体领域展现出强大的应用前景,为癌症治疗提供了新的策略。
02
代表性双抗药物
2.1 Hemlibra(艾美赛珠单抗)
Hemlibra是日本中外制药株式会社(Chugai Pharmaceutical)发现,并由中外制药和罗氏共同开发由罗氏研发的一种双特异性因子IXa和因子X定向抗体,可桥接活化的 IX 因子和 X 因子以替换缺失的活化因子 VIII 的功能,恢复A型血友病患者的凝血过程(图4)[3]。
Hemlibra于2017年11月首次被FDA批准上市并于2018年被FDA扩增新适应症,适用于成人和儿童血友病A患者,无论是否伴有因子VIII抑制剂,用于预防或减少出血事件的发生。
Hemlibra是近20年来FDA批准的首个用于治疗A型血友病的创新药物,凭借其便捷的给药方式和良好的疗效,在血友病A治疗领域占据重要地位。
2018年11月,Hemlibra在中国获批,成为国内首款上市的双特异性抗体药物。截至2024年,Hemlibra已在全球100多个国家获批用于治疗体内存在凝血因子VIII抑制物的A型血友病患者,并在80多个国家获批用于体内不存在凝血因子VIII抑制物的患者。
Hemlibra的研发是迄今为止最大的A型血友病临床研究项目之一,包括多个关键性III期HAVEN研究(HAVEN-1、HAVEN-2、HAVEN-3、HAVEN-4)。这些研究验证了Hemlibra在不同患者群体中的安全性和有效性,包括存在或不存在凝血因子VIII抑制物的患者。
Hemlibra自2017年上市后销售额逐年上升,并于2020年成功突破20亿美元销售额大关,成为重磅品种。2021年,Hemlibra全球销售额为30.88亿瑞士法郎(约34.34亿美元,按当下1瑞士法郎约等于1.112美元,下同),同比增长41%;2022年Hemlibra销售额为38.23亿瑞士法郎(约42.51亿美元),同比增加27%,2023年销售额达到41.47亿瑞士法郎(约49.3亿美元),2024年Hemlibra销售额达到45.03亿瑞士法郎,同比增长12%(图5)[4]。
2.2 Vabysmo(Faricimab)
Vabysmo (Faricimab)是由罗氏开发的第一个获批用于眼部疾病的双特异性抗体,作用于血管生成素-2(Ang-2)和血管内皮生长因子-A(VEGF-A),Ang-2和VEGF-A通过破坏血管稳定、导致新的渗漏血管形成和增加炎症,从而导致视力丧失。
通过阻断涉及 Ang-2 和 VEGF-A 的通路,Vabysmo 可以抑制许多威胁视力的视网膜疾病相关的两条信号通路(图6)。
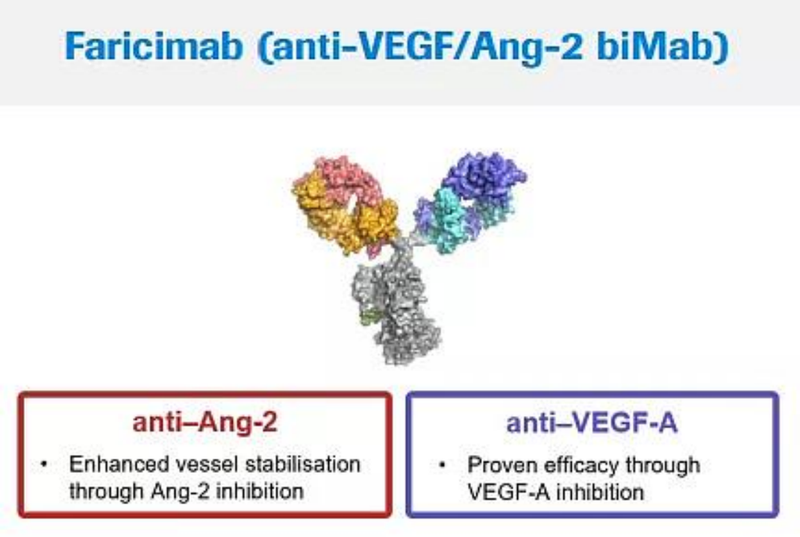
Vabysmo 在全球100多个国家获得批准,包括美国、日本、英国和欧盟等,用于治疗新生血管性或“湿性”年龄相关性黄斑变性和糖尿病性黄斑水肿患者等。
2022年1月,Vabysmo首次获 FDA 批准上市,获批适应症有湿性年龄相关性黄斑变性(wAMD)和糖尿病黄斑水肿(DME)。
2023年10月27日,FDA批准Vabysmo用于治疗视网膜静脉阻塞(RVO)患者,成为继wAMD和DME之后第三项获批适应症。
2024年7月,罗氏公布了Vabysmo治疗DME的RHONE-X四年临床试验数据。结果显示,Vabysmo在接受长达四年治疗的DME患者中耐受性良好,超过90%的患者达到DME症状消失的标准。
在该研究中,近80%的患者将治疗间隔延长至每三个月或四个月一次,同时保持了视力改善。
2024年ARVO会议上,在对关键性TENAYA(NCT03823287) 和 LUCERNE (NCT03823300)试验的事后分析中,Philip P Storey 医学博士提供的结果证明了Vabysmo延长的治疗结果,以及其将剂量减少到每20周一次的潜力。
这项为期两年的研究在 wAMD 患者中比较了Vabysmo与Eylea(阿柏西普)(每 8 周给药一次),结果显示近80%的Vabysmo患者实现了12周或16周的给药间隔。
此外,该研究确定,56%每16周给药一次的患者可以使用相同的标准延长至每20周给药一次,而44%的患者由于解剖变化(例如中央子视野厚度增加)不符合这种延长的条件[5]。
Elevatum(NCT05224102)是一项为期 1 年的4 期、多中心、开放标签、单臂、1 年试验,包括黑人或非裔美国人的患者(占总入组人数的 ~45%);西班牙裔或拉丁美洲人(占总入学人数的 ~45%);或美洲原住民、阿拉斯加原住民、夏威夷原住民或太平洋岛民(占总入学人数的 ~10%)。将从 47 个地点招募美国和肯尼亚的大约 120 名未接受过治疗的 DME 患者(年龄≥ 18 岁)。
在ELEVATUM 4期研究中,Vabysmo在种族和民族代表性不足的DME患者群体中显示出显著的视力改善,平均视力表改善12.3个字母(约2.5行),这一效果在不同种族和民族群体中保持一致[6]。
2024年12月13日,罗氏宣布欧洲药品管理局已批准 Vabysmo(faricimab) 6.0 mg 单剂量预充针(PFS)用于治疗nAMD、DME和RVO后黄斑水肿。这三种疾病共同影响了欧盟(EU)的900多万人。
Vabysmo PFS 是第一个也是唯一一个含有双特异性抗体的预充针,为目前可用的 Vabysmo 西林瓶提供了一种方便的替代品[7]。
Vabysmo自上市以来,销售额一直表现不错,2022年它的全球销售额达到5.91亿瑞士法郎(6.6亿美元),2023年全球销售额达到23.6亿瑞士法郎(26.34亿美元),2024年全球销售额高达38.64亿瑞士法郎(约46.0亿美元)(图7)[4]。
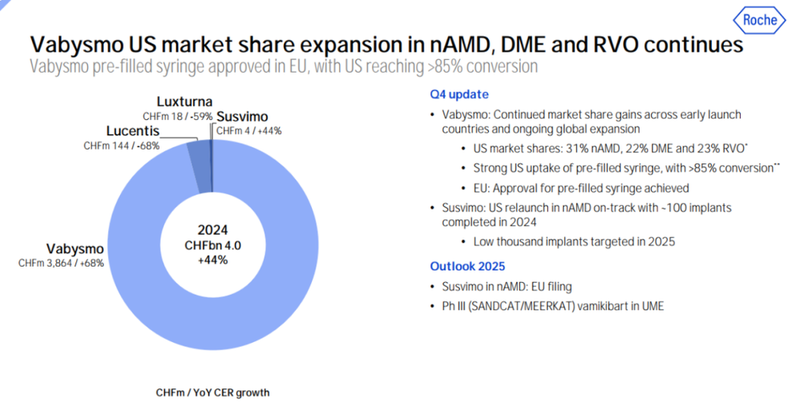
图7. Vabysmo近两年各个季度销售额
2.3 Blincyto(博纳吐单抗)
Blincyto是由安进研发的全球首个CD19-CD3双特异性T细胞接合器(BiTE)药物,由两个主要片段抗CD3 scFv(VL-VH)和抗CD19 scFv(VL-VH)组成,于2014年12月3日获FDA批准上市,用于治疗复发的费城染色体阴性的B细胞急性淋巴性白血病(B-ALL)。
2022年12月,Mark等人在ASH会议上报道了Blincyto的ECOG-ACRIN E1910 3期试验结果:中位随访时间为43个月时,Blincyto组和化疗组中位总生存期(OS)分别为未达到和71.4个月(HR=0.42,95%CI:0.24 -0.75)(图8)[8]。
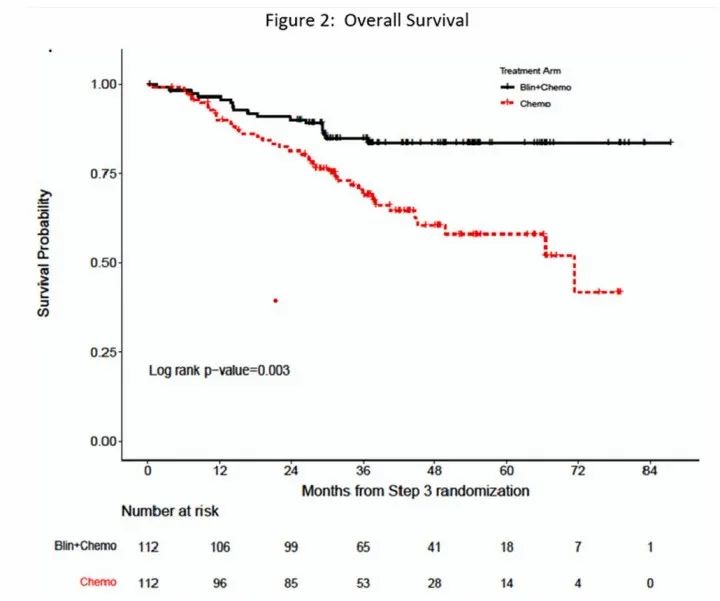
图8. ECOG-ACRIN E1910 3期试验结果
2024年6月14日,安进宣布FDA已批准Blincyto 用于治疗巩固期 CD19 阳性费城染色体阴性 B 细胞前体急性淋巴细胞白血病 (B-ALL) 的成人和儿童一个月或一个月以上的患者,无论可测量的残留疾病 (MRD) 状态如何。
此次批准是 Blincyto的第三个适应症,主要基于 ECOG-ACRIN 癌症研究组领导的 3 期 E1910 临床试验,该试验研究了接受诱导后巩固治疗的新诊断的费城染色体阴性 B 细胞淋巴细胞淋巴瘤患者,旨在深化缓解以获得持久反应。
研究结果表明,与单纯化疗相比,Blincyto加入多期巩固化疗显示出更好的OS。Blincyto联合化疗组 (n=112) 的3年OS为84.8%,化疗组 (n=112) 的 3 年OS为69%,OS风险比为0.42。中位随访期为4.5年,Blincyto联合化疗组的5年OS为82.4%,化疗组为62.5%。
2025年1月29日,欧盟委员会基于ECOG-ACRIN癌症研究组主导的3期E1910临床试验结果批准Blincyto用于新诊断的CD19阳性、费城染色体阴性B细胞前体急性淋巴细胞白血病(B-ALL)成年患者的巩固治疗。
Blincyto自上市后销售额稳步增长,2021年达到4.72亿美元,然后可能由于Blincyto半衰期仅 2 小时,患者需要频繁给药,以及其它药物的竞争导致销售额在2022年下滑至3.36亿美元,不过由于BB-ALL患者的广泛处方销售额得到剧增,2023年Blincyto销售额达到8.61亿美元,2024年第四季度销售额为3.81亿美元,全年销售额达到15.9亿美元(图9)。
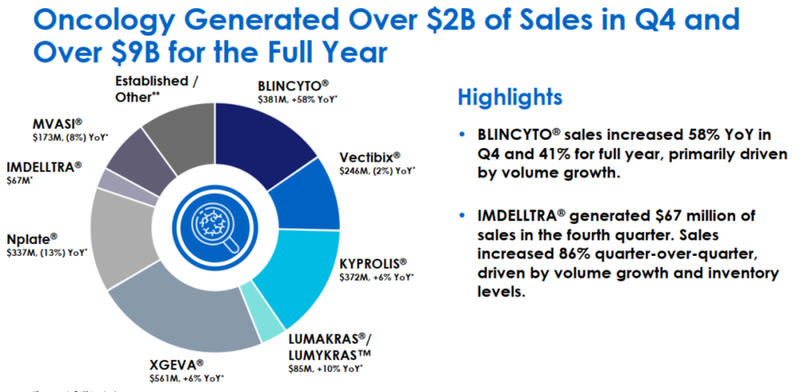
2.4 卡妥索单抗(Catumaxomab)
卡妥索单抗是首个获批的靶向CD3和EpCAM的双特异性抗体,是一种三单抗(Triomab)形式,它具有两个特异性抗原结合位点和一个功能性Fc结构域,能够同时靶向肿瘤细胞上的上皮细胞黏附分子(EpCAM)和T细胞上的CD3分子(图10)。
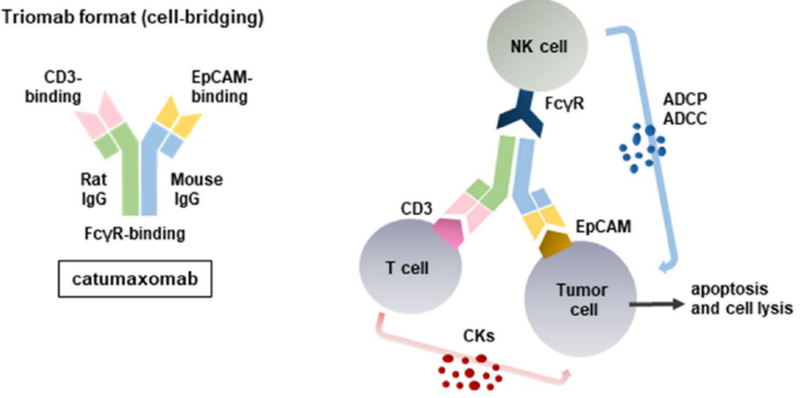
卡妥索单抗于2009年获得欧盟(EMA)批准上市,用于治疗恶性腹水。但是卡妥索单抗上市后市场表现不佳。从2009年至2013年,卡妥索单抗的全球销售额持续低迷,分别为166万、332万、443万、454万和270万美元。这种持续的市场表现不佳最终在2017年6月正式退市。
尽管如此,卡妥索单抗在临床试验中展现出的疗效并不差,尤其是在治疗因胃癌导致的恶性腹水方面表现出色。
基于此,凌腾医药重启了卡妥索单抗在国内的开发。2020年7月,卡妥索单抗开展了伴腹膜转移胃癌的国际多中心三期临床试验。2021年4月,卡妥索双抗获得国家药品监督管理局批准的一项用于治疗非肌层浸润性膀胱癌(NMIBC)的1/2期( NCT04799847)的临床试验许可。
2022年11月,凌腾医药欧洲合作伙伴Lindis Biotech 在14届欧洲泌尿系统癌症多学科大会(EMUC)上公布了卡妥索单抗CATUNIBLA I期临床试验最新结果:卡妥索单抗在NMIBC中显示良好的安全性和初步疗效。
2025年2月14日,Lindis Biotech递交的卡妥索单抗上市许可申请(MAA)已获得欧洲委员会(EC)批准,用于腹腔内治疗EpCAM阳性且不适合进一步全身抗肿瘤治疗的成人患者的恶性腹水。
这标志着卡妥索单抗在全球范围内重新获得认可,并为恶性腹水患者带来了新的治疗希望。
2.5 开坦尼和依达方
中国双抗市场竞争激烈,康方生物凭借开坦尼(卡度尼利单抗)和依方达(依沃西单抗)等产品在肿瘤免疫双抗领域占据重要地位。
开坦尼是由康方生物研发的靶向PD-1和CTLA-4 的一种对称的四价双特异性抗体,具有可结晶片段(Fc),于2022年6 月 29 日首次在国内获批上市,用于既往接受过含铂化疗治疗失败的复发或转移性宫颈癌患者(图11)。
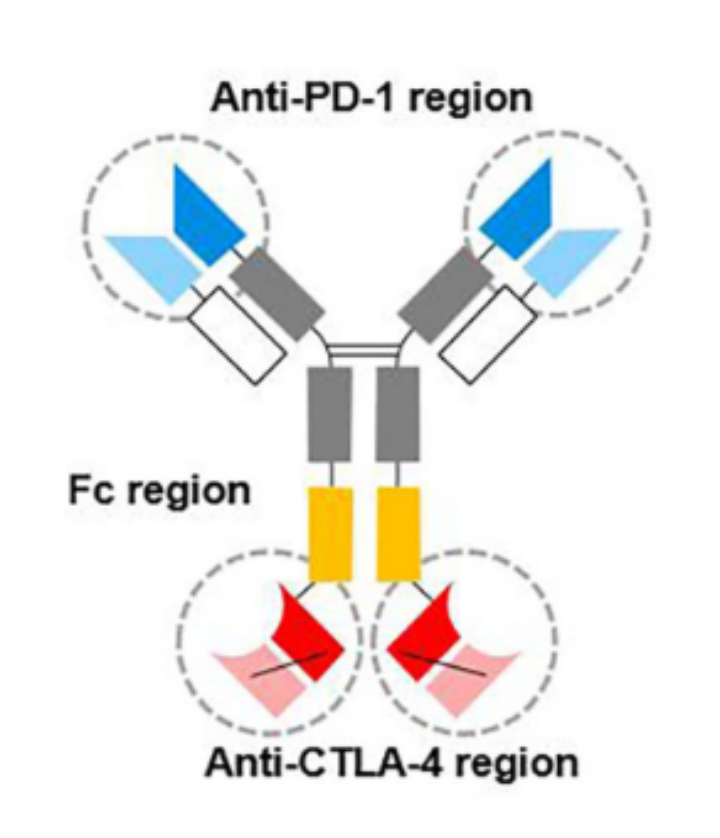
开坦尼是中国自主研发的首款双特异性抗体药物,也是全球首个获批的PD-1/CTLA-4 免疫双抗,具有里程碑式的意义。
2024年9月,开坦尼联合含铂化疗方案获批用于局部晚期不可切除或转移性胃或胃食管结合部腺癌患者的一线治疗。
2024年9月,开坦尼联合方案一线治疗晚期胃癌全人群适应症已获国家药品监督管理局(NMPA)批准上市。
该适应症的获批是基于Ⅲ期COMPASSION-15(AK104-302)研究的优异阳性结果:无论PD-L1表达水平如何,开坦尼联合方案相较于化疗能显著延长患者的总生存期并降低死亡风险,对于肿瘤治疗的客观缓解和远期生存获益尤为显著[9]。
开坦尼已被纳入多个临床治疗指南,包括《CSCO免疫检查点抑制剂临床应用指南(2024版)》、《基于PD-L1蛋白表达水平的胃癌免疫治疗专家共识(2023年版)》等,2024年11月,开坦尼被纳入《CACA胃癌整合诊治指南2024版》,成为唯一不限PD-L1表达的优先推荐肿瘤免疫治疗药物。
依达方是由康方生物自主研发的全球首创PD-1/VEGF双特异性肿瘤免疫治疗药物。这是一种四价双特异性抗体,融合了抗PD-1单抗的单链可变片段(scFv)和抗VEGF单抗的完整结构。
依达方于2024年5月获得NMPA批准上市,用于治疗EGFR-TKI治疗失败后的局部晚期或转移性非鳞状非小细胞肺癌(nsq-NSCLC)。
依达方是全球第一个获批上市的“肿瘤免疫+抗血管生成”机制的双特异性抗体新药,也是中国第二个获批上市独立自主的双特异性抗体新药[10]。
依达方获批是基于一项在中国开展的随机、双盲、多中心III期临床研究(AK112-301/HARMONi-A)结果。
研究结果显示:依达方联合化疗组的中位PFS为7.1个月,显著优于对照组的4.8个月(HR=0.46,P<0.001),降低了54%的疾病进展或死亡风险;数据成熟度达到52%时,依沃西单抗联合化疗组的中位OS为17.1个月,对照组为14.5个月,降低了20%的死亡风险[11]。
目前,依达方还在进行多项全球多中心临床研究,包括一线治疗鳞状非小细胞肺癌(sqNSCLC)的3期研究以及与化疗联合治疗EGFR突变非鳞状NSCLC的全球多中心3期研究等。
令人欣喜的是依达方已获得海外授权。康方生物与Summit Therapeutics达成合作,授予其在美国、加拿大、欧洲、日本等地区的开发和商业化权利。Summit也找到辉瑞,与后者的ADC药物开展联合用药研究,不断放大这款双抗药物的潜力。
此外,国产双抗出海交易在2024年呈现大爆发态势,如同润生物与默沙东达成的CD3/CD19双抗交易,金额高达13亿美元。
小结
双抗技术得到快速发展,从早期的主要通过化学偶联或杂交杂交瘤技术制备到现在发展了很多新的技术,如BiTEs、DART和TandAb等,双抗药物的稳定性和活性也得到了大幅度提升。
双抗药物的市场规模也在快速发展,根据市场分析,预计从2024年到2033年,全球双抗药物市场将以37.5%的复合年增长率增长,到2033年市场规模将达到约1926亿美元。

公众号内回复“ADC”或扫描下方图片中的二维码免费下载《抗体偶联药物:从基础到临床》的PDF格式电子书!
公众号已建立“小药说药专业交流群”微信行业交流群以及读者交流群,扫描下方小编二维码加入,入行业群请主动告知姓名、工作单位和职务。
