
骨形成蛋白受体受体 2(BMPR2)的杂合突变存在于15%-40%的特发性肺动脉高压(PAH)中,在非遗传性 PAH 中观察到BMPR2蛋白表达降低。大多数 BMPR2突变会导致单倍体不足和信号转导减少。这种减少驱动肺动脉、内皮细胞和平滑肌细胞的增殖和细胞凋亡抵抗,这会导致肺血管阻力增加、右心衰竭和死亡。因此,增强BMP信号转导可能为治疗PAH提供一种变革性的、新颖的方法。
有研究表明,SMAD泛素化调控因子1(SMURF1)作为一种负向调控BMP信号通路的关键E3连接酶,在PAH患者中表达显著上调,并通过泛素化降解BMPR2和SMAD1/5/8,驱动血管平滑肌细胞增殖和抗凋亡。实验表明,SMURF1抑制剂可恢复BMP信号通路,改善肺血管细胞稳态,逆转动物模型的病理改变。而HECT型E3泛素连接酶作为泛素化系统的核心成员,通过催化泛素从E2转移到靶蛋白,调控蛋白质的降解、定位及活性。然而,与传统的E3连接酶不同,HECT家族成员的活性位点暴露于蛋白表面,缺乏传统意义上的“活性口袋”,阻碍了识别抑制剂的设计。
基于这一机制,本研究通过机器学习辅助的虚拟筛选,发现了一种针对HECT类E3泛素连接酶(其中E6AP是该类的原型成员)的抑制剂,并证实了其依赖甘氨酸铰链的变构调控机制。该研究为靶向 HECT 及其他含甘氨酸铰链的蛋白质开辟了新的药物开发空间。
1. 抑制剂作用机制
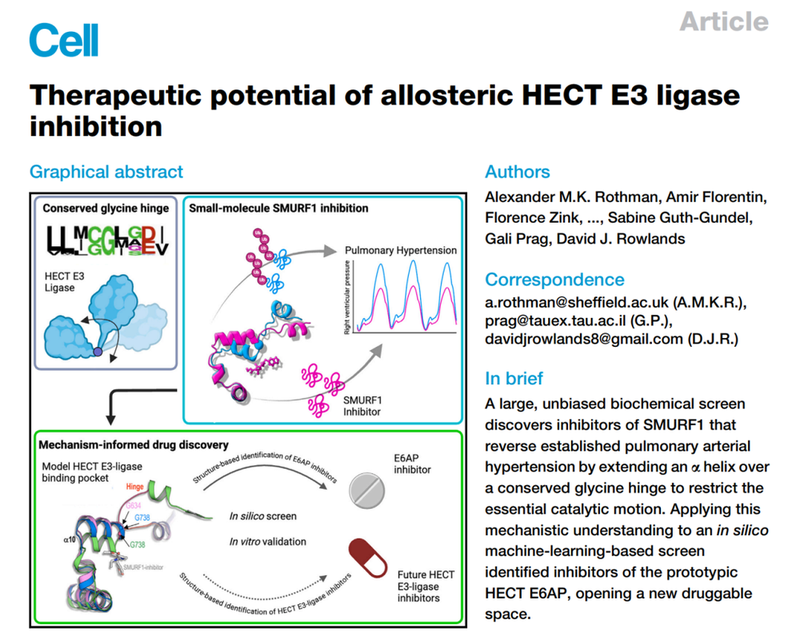
通过X射线晶体学检测,研究者发现这些抑制剂通过结合HECT结构域N叶和C叶之间的隐秘腔,延伸αH10覆盖G634铰链,限制其灵活性。所有家族成员叠加的HECT结构域模型表明,N叶和C叶围绕该铰链区域的相对运动,对泛素通过HECT C叶从E2酶向靶蛋白的转移过程具有重要作用。同时,SMURF2与Cpd-8复合物的结构显示出它与SMURF1抑制剂复合物具有相同的隐腔结合,但是抑制剂的结合并没有延长SMURF2的αH10,也并没有降低SMURF2的活性。基于这些结构,研究人员假设SMURF1 活性受到抑制是因为抑制剂的变构活性诱导使得甘氨酸铰链的长度和柔韧性减少。
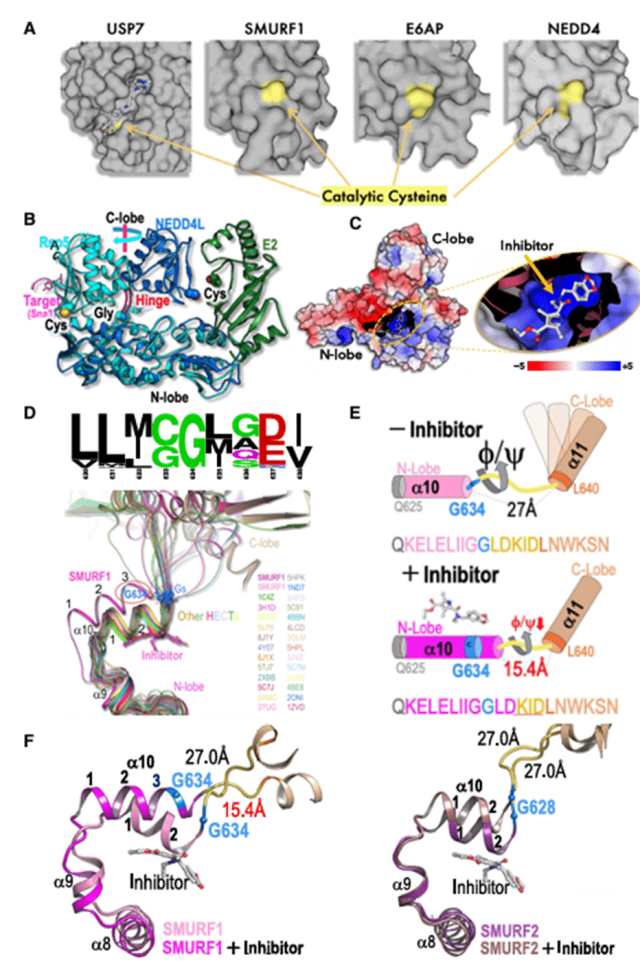
图1.SMURF1和SMURF2 HECT 结构域与抑制剂复合物
2. 逃逸突变体验证
首先,为了研究铰链长度和灵活性对于SMURF1功能的重要性,研究者构建了两类突变体:(1)铰链灵活性降低的突变体,通过用脯氨酸(G634P)取代保守的甘氨酸而产生(2)铰链长度缩短的突变体,通过删除三个氨基酸残基(D637KID)产生。两种突变体都降低了泛素化活性,证实了甘氨酸铰链灵活性和长度的关键作用。
研究表明,Cpd-8能够显著减弱SMURF1 的活性。因此,研究者构建了两个 SMURF1 突变体,插入GGLD(使铰链灵活性降低)或引入SMURF2突变(G633C/D636G,使铰链长度缩短),使SMURF1抵抗抑制剂作用。在没有抑制剂的情况下,637GGLDINS和G633C/D636G 双突变体均未显著改变 SMURF1 活性。在 Cpd-8 存在下,两种突变体都保留了超过 75% 的活性,表明保持甘氨酸铰链的柔韧性和长度可以有效地逃逸抑制剂。
此外,分子结构分析表明,SMURF1中D636、R686和N507之间形成的非共价键网络能够稳定化合物结合诱导产生的变构延伸态αH10。D636G、R686A和N507A单点突变虽未显著改变酶活性,但均使SMURF1对Cpd-8的抑制作用产生抗性,证实了aH10的构象稳定对抑制剂发挥效力的关键作用。为验证该稳定网络的引入能否使SMURF2获得抑制敏感性,研究者构建了G630D突变型SMURF2。该突变体对磷酸化模拟型SMAD1底物肽段的催化活性未发生显著改变,但表现出对Cpd-8的敏感性。上述结果共同表明:抑制剂通过结合SMURF1诱导αH10绕保守甘氨酸铰链发生延伸,这种构象变化由非共价键网络稳定,进而限制催化功能必需的甘氨酸铰链的柔韧性和延展度。而SMURF2的抑制抗性源于G630残基的存在,该位点在化合物存在时无法形成维持aH10延伸态所需的键合网络。
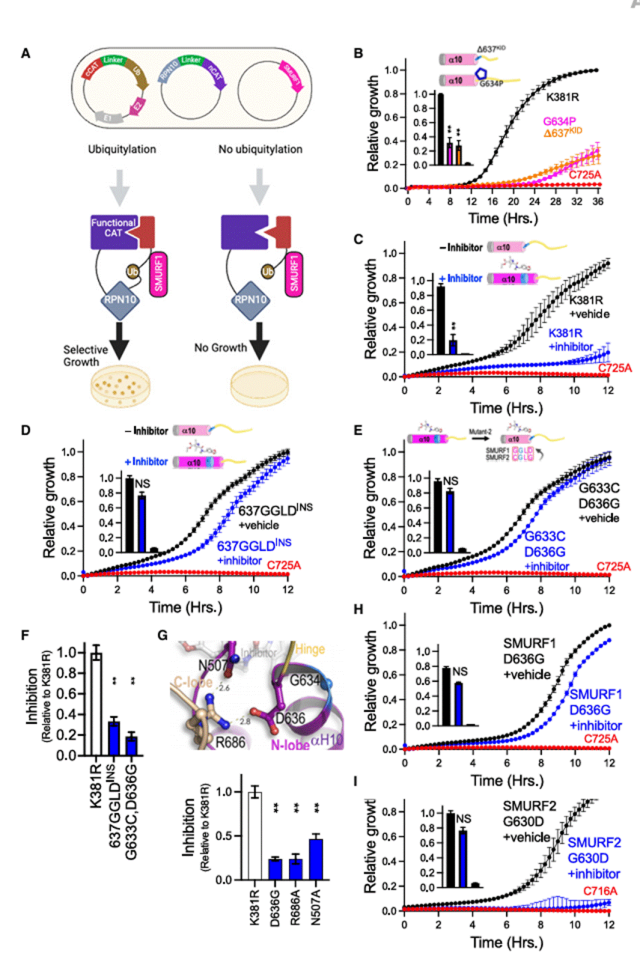
图2.SMURF1 的变构抑制。
3. SMURF1在PAH中的病理作用
研究表明,PAH患者肺血管内皮/平滑肌细胞SMURF1显著升高,PAH发病与BMP信号减弱密切相关,SMURF1作为该通路的关键负调控因子,其表达水平受BMP配体(如BMP4)动态调节。缺氧及TGF-β信号可显著上调SMURF1,加剧血管重塑。
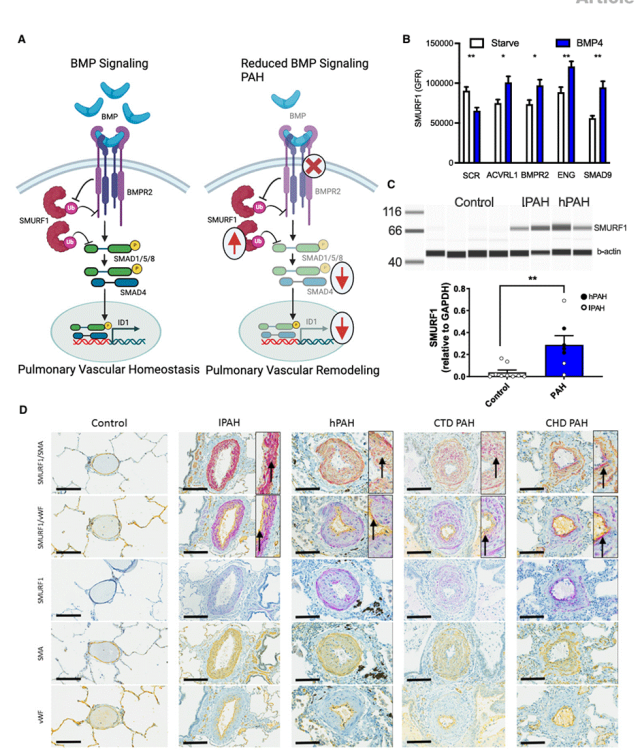
图3.SMURF1 在 PAH 中的表达。
4. 靶点验证与信号恢复
研究证实,SMURF1作为E3泛素连接酶,可不依赖SMAD6/7等适配蛋白,直接通过其WW结构域结合BMPR2受体并催化其泛素化降解,同时靶向磷酸化SMAD1阻断BMP信号传导,形成对通路的双重抑制。实验证实,SMURF1的催化活性受甘氨酸铰链构象动态调控:铰链柔韧性降低(如G634P突变)增强泛素化效率,而维持灵活性的突变(如G633C/D636G)可逃逸抑制。在疾病模型中,变构抑制剂的应用显著逆转这一病理过程——PAH患者来源的肺动脉平滑肌细胞(PASMCs)和内皮细胞(PAECs)中,BMPR2/SMAD1蛋白水平及ID1表达显著升高,并激活BMP反应元件,恢复信号传导功能。功能实验进一步显示,抑制剂在低浓度BMP9下减少内皮细胞凋亡与增殖,在BMP4存在时抑制平滑肌细胞增殖迁移,且疗效不受BMPR2突变影响。蛋白质组学分析表明,SMURF1抑制通过上调RhoA、TGFBR1等靶蛋白及BMP通路分子,协同调控TGF-β/IL-1β信号、细胞外基质重塑等病理进程。该研究从分子机制(泛素化双靶点抑制)到细胞功能(稳态恢复)多维度阐明SMURF1变构抑制的治疗潜力,为肺动脉高压的精准干预提供了创新策略。
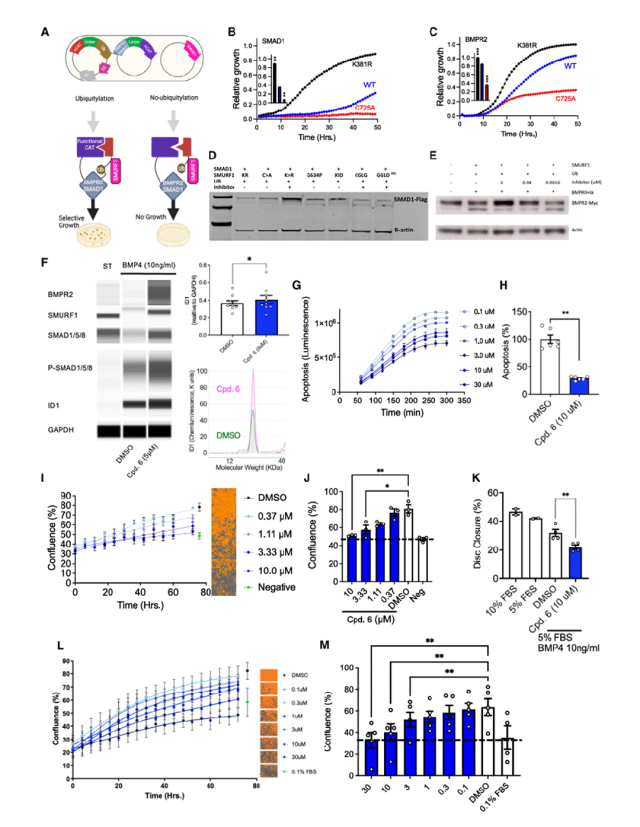
图4.SMURF1 抑制剂恢复 BMP 信号传导和肺血管细胞稳态
5. 体内疗效与安全性
在野百合碱和SU5416+缺氧诱导的PAH大鼠模型中,SMURF1抑制剂显著降低了右心室收缩压,减少了肺小动脉的肌化程度。这证明了SMURF1抑制剂能够逆转肺动脉高压。同时,为了评估变构SMURF1 抑制剂的潜在毒性,研究者对 MCT 研究中的血液和器官进行了详细的生化和组织病理学评估,未发现显著毒性。
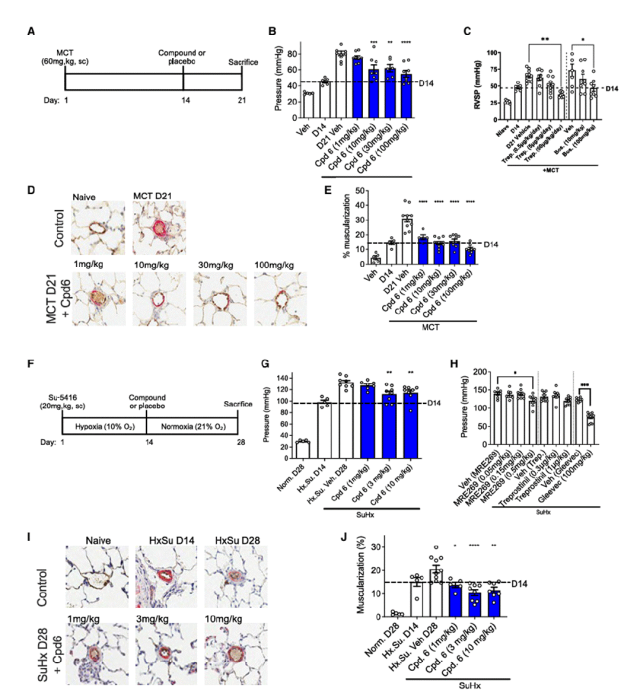
图 5.抑制SMURF1 治疗实验性 PAH
6. 机制拓展至E6AP
基于SMURF1变构机制,通过机器学习筛选发现了E6AP抑制剂i-27。实验表明,i-27能够通过稳定E6AP的抑制构象,限制甘氨酸铰链的灵活性,从而抑制E6AP活性。该发现可能为发现其他变构抑制剂(包括 HECT连接酶)提供可用策略。
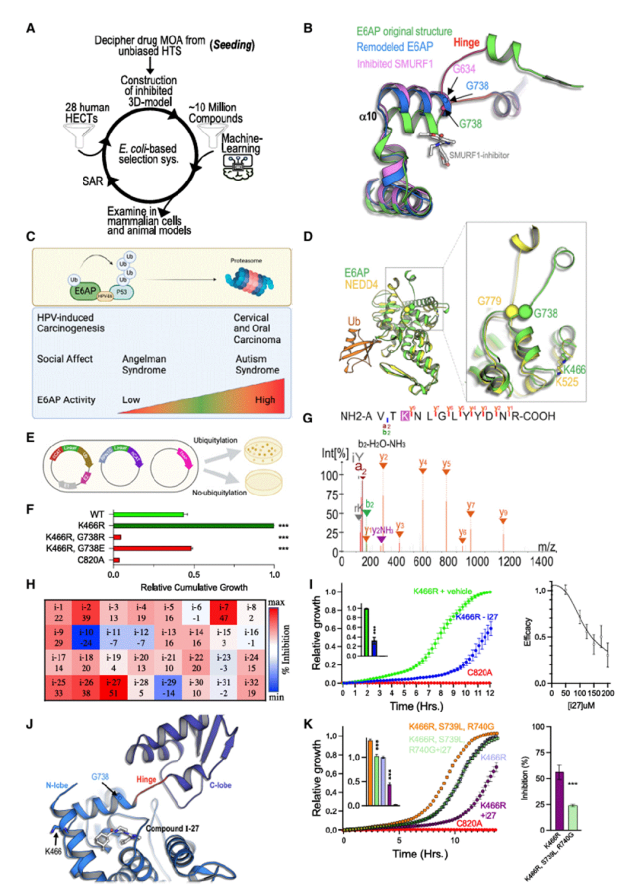
图 6.基于结构的 E6AP抑制剂鉴定
在肺动脉高压动物模型中,SMURF1抑制剂可恢复BMP信号,改善血管细胞功能,甚至逆转疾病进展,显示出明确的治疗潜力。进一步地,研究团队基于该变构机制,结合机器学习开展虚拟筛选,成功筛得并验证了E6AP(HECT 类原型酶)的甘氨酸铰链依赖性抑制剂。这一发现不仅为SMURF1和E6AP等泛素连接酶的靶向治疗提供新策略,也首次系统性揭示了“甘氨酸铰链”作为药物作用位点的可行性,拓展了变构药物设计的新空间。
